Is there another way to measure the benefit from any medication?
We all want the cure, the Complete Response (CR) that can lasts many months or years. Often we have to settle for some reduction in our tumors or mets, a Partial Response (PR). But even “Stable Disease” is welcome news. To get that cancer back in its cage, even for a time, is better than “Progressive Disease”. When the cancer is progressing, your life may be regressing, and that isn’t what you want to hear. That Progression Free Survival (PFS) has to start with stopping the cancer.
As complete and durable (ten years) responder to high dose interleukin 2 (HD IL-2), I welcome any discussions of “Clinical Benefit (CB)”. CB includes all the good responses with any cancer treatment, CRs, PRs, and SDs. We and our doctors need this information to make informed decisions about treatment, for IL2 or other meds. The value of Stable Disease has been ignored in many studies. Maybe there are lessons here for you and your doctors, especially about the under-utilized HD IL2.
Clinical benefit (CB) of high-dose interleukin-2 (HD IL-2) in clear cell (cc) metastatic renal cell carcinoma (mRCC).
There are few new studies about the use of HD IL2 following the approval of the targeted therapies. The ease of use of these agents, along with the desire not to send patients to specialty centers for IL2, limited its use. It was difficult to select patients, and the CR and PRs were relatively small in number. Doctors often did not discuss the possibility of a cure with their patients. Did patients also miss the chance for Stable Disease, and with it, a “Clinical Benefit”?
Patients in this study who did not have a CR, but whose cancer stopped growing benefited. That CB was not counted in terms of the approval of the drug, nor do doctors consider it in their recommendations. Should this possibility be discussed with patients? Most patients would surely answer, “Yes!” to that question.
The researchers recognize of the value of Stable Disease (SD) as an outcome, versus only Complete Response (CR) or Partial Response (PR). The usual outcome measures, Progressive Free Survival (PFS), or Overall Survival (OS), are noted, as isTime to Next Treatment (TNT). TNT implicitly recognizes that a failed or limited response will likely be followed by another treatment. Early on, there were no subsequent treatments, sad to say.
The original clinical trial which led to FDA approval of HD IL2 recognized only CR, which was 5%, with the median not reached during the trial, and PR, which was 14%. Study footnotes indicate that three of the PRs had surgery which rendered them disease free at the time of the publication. This would now be called a “salvage therapy”, and put them in the No Evidence of Disease (NED) class. A different analysis of this data would have upped the CRs some small percentage, and some SD would also have been found.
Also the definition of PR was 50% or greater reduction in measurable tumor size, the sum of the perpendicular diameters of all lesions, with no new increase of sizeof any other mets. Far less strict measurements of PR were used in the targeted therapy trials, with a 30% tumor reduction defined as a Partial Response.
With those definitions in mind, note that there are CRs in 11% of patients, with a PR in an additional 6% of patients. Most important is the SD category, which was achieved for 31% of all patients. This total of 47% is described for the group as being of Clinical Benefit (CB). Certainly patients value the responses of SD, which seems to have provided slightly over one year versus 3-4 months benefit to those who did not have SD.
When comparing the value of Objective Response (OR) with its median of 1616 days to that of Stable Disease (SD) measured as 1476 days, one can clearly see the value of achieving Stable Disease. Unfortunately, those patients with Progressive Disease, or with responses Not Evaluable (NE), showed OS of 365 days.
Patients should be aware of these definitions and the impact the lack of parallel comparisons in making these critical decisions. Ten years ago, the patients reminded one another to stay alive until the next treatment. Having Stable Disease made that possible. Let’s apply the same tests to all the available treatments when making these life-changing choices of treatment.
Author(s): Neeraj Agarwal, David D. Stenehjem et al University of Utah, Huntsman Cancer Institute, Salt Lake City, UT; Comprehensive Cancer Centers of Nevada, Las Vegas, NV; Pharmacotherapy Outcomes Research Center, College of Pharmacy, University of Utah, Salt Lake City, UT
Background: HD IL-2, an immunotherapy, is a standard of care for a select group of patients (pts) with mRCC. Generally objective response (OR) rates, i.e. complete response (CR) + partial response (PR), of 16-20% are discussed with pts, but not disease stabilization (SD). Recent data suggest that cancer immunotherapy may improve survival without inducing OR. Thus, treatment with HD IL-2 may provide survival benefit to an additional group of pts not experiencing OR, but only SD as the best response. Here we report CB (OR+SD), and specifically report outcomes of cc mRCC pts experiencing SD as the best response, on treatment with HD IL-2.
Methods: All sequential cc mRCC pts treated with HD IL-2 at the University of Utah Huntsman Cancer Institute from 2000-2012 were included. Pts were evaluated for best response, progression-free survival (PFS), time to next treatment (TNT) and overall survival (OS). Two practitioners independently reviewed HD IL-2 response with discrepancies adjudicated by a third reviewer.
Results: 85 pts, 79% male, were identified with a median age of 56 (range 32-76) years. Pts belonged to the following MSKCC risk categories: 11 (13%) good, 70 (82%) intermediate, and 4 (5%) poor risk. A CR was identified in 9 (11%), PR in 5 (6%), SD in 26 (31%), progressive disease (PD) in 38 (45%), and unknown/not evaluable (NE) in 7 (8%) pts; yielding a clinical benefit in 40 (47%) pts. The median PFS, TNT, and OS in these individual groups of pts are compared in the table.
Conclusions: A clinical benefit of HD IL-2 was achieved in nearly half of all clear cell mRCC patients. OS was not significantly different in OR and SD groups. Even though OR favorably determine outcomes, SD is also an important response criterion, and may be discussed during counseling patients for treatment with HD IL-2.
PFS, days
TNT, days
OS, days
Overall
152
264
817
SD vs PD and NE
337 vs 78 (p<.0001)
373 vs 110 (p=.0001)
1,476 vs 365 (p=.0003)
CB vs PD and NE
791 vs 78 (p<.0001)
735 vs 110 (p<.0001)
1,616 vs 365 (p<.0001)
OR vs SD, PD and NE
NA vs 99 (p=.0003)
953 vs 166 (p<.0001)
1,616 vs 603 (p=.0021)
OR vs SD
NA vs 337 (p=.0234)
953 vs 373 (p=.0015)
1,616 vs 1,476 (p=.2094)
Abbreviation:PFS, Progression Free Survival; TNT, Time to Next Treatment, OS, Overall Survival; NA, not achieved;SD, Stable Disease; PD, Progressive Disease; NE, Not Evaluable; CB, Clinical Benefit;CR, Complete Response; PR, Partial Response;OR, Objective Response
“Now what?” may be the first coherent question a newly diagnosed cancer patient asks. Maybe the smarter version of that is “What–when and why?” And your doctor had better have a good answer, as to the treatment, the when and the why.
We cancer patients usually get surgery “first”, even when the disease has spread. Primary surgical strike and then a clean-up operation, in the ‘war on cancer’ parlance, we think–when we can think. “But which is the best and first clean-up approach?” we must ask. “What works the best? What can I take with my other health problems? Where does surgery or radiation fit in this scheme? What does the doctor favor and why? Where do I get this treatment? And then what?”
Treatments and their sequence are often chosen with little reliance or clarity as to the data. But some light was shed today at ASCO (American Society of Clinical Oncology). It released a comparison of the sequencing of High Dose Interleukin2 (HD IL2) and of targeted therapies for metastatic RCC. Which should come first?
It shouldn’t be a high-stakes gamble to choose a medication, as no one can guarantee any results–with any of the meds. You take a chance with any drug, so which do you start wi We may be closer to a logical approach in sequencing these drugs. Sequencing of these highly different medications has measurable effect on overall survival (OS)—and to patients’ lives. That sequencing is critical and certainly can extend life, even when treatments fail, as they so often do.
A retrospective study of 97 US patients who received HD IL2, before or after a targeted therapy was just presented at ASCO. These patients were followed for a median duration 37 months–half more than 37 months, half fewer than 37 months. Of that group, 22% had either a partial (14%) or complete (8%) response to HD IL2. (No specifics as to what was a “partial” response, perhaps a 30% shrinkage of the total tumor burden). In addition, another 24% of patients had Stable Disease(SD). Thus, nearly half of these patients benefited from having had HD IL2.
Stable disease is better than progressive disease, as any patient knows, though it was rarely measured in older trials. Though we patients really want a cure, we do want to be around for the next treatment, to have a surgery or ablation to remove the “stable” tumor, or to try another medication.
Of these 97 patients, 82 received HD IL2 before any targeted therapy. Another 15 patients had HD IL2 following a TKI therapy. That timing made an important difference. HD IL2 followed by the TKI, showed a median Overall Survival (OS) of 61.8 months. The OS of those with the TKI before the HD IL2 was 48 months. A median, not an average, so half lived longer, half lived shorter than the quoted medians.
A pre-2006 NCI (National Cancer Institute) series showed a 19 month median survival for HD IL2 alone, and a similar 19 months for the use of targeted therapy alone. Using the two in sequence dramatically improved OS, especially when HD IL2 was first line of treatment. Obviously things have improved, though it can be very difficult to compare older trial data, as so many variables are different–including the type of RCC the patients had as they entered the trials.
Several points can be made from this study. First, no therapy should be examined only as to Complete or Partial Response. Stable Disease also adds to Overall Stability. To stop the tumor from growing, even if for a period of time, is valuable to patients and can prep them for the next anticipated treatment. Sure beats tumor growth!
Second, therapies should be chosen to maximize their impact on the overall survival of the patient. Some patients will naturally be precluded (or delayed) from surgery, or taking one drug due to existing co-morbidities, due to heart disease or liver damage. For those post-op patients, likely to tolerate the side effects of HD IL2, it should be given in a first-line setting.
The most critical variables that impact patients are the recommendations and expectations of the physician. Most patients are not even told about HD IL2 treatment, or it is dismissed casually as “not for you”. Others are told to wait until more mets emerge, with some weird theory that waiting for more trouble is a good thing! Many nephrectomy patients are not monitored post-operatively, despite the risk of mets. This is surely an indicator of the lack of knowledge by urologists. Still others are told that the disease has spread, and that nothing can be done–also untrue.
The rarity of RCC and its variants leaves most physicians unaware of all options in the field, and how to any one might suit for a particular patient. Most oncologists to whom patients are referred, have little or no experience treatmenting for RCC, or may not access to academic centers for support until it is too late. Even the pathology of the primary tumor and later metastases may be questionable, adding to the challenge of care.
With the dramatic changes in the RCC field, this is to be expected—but not tolerated. The patient may have to provide his physician with the data that can extend or save his life, which is a sad but realistic commentary on the field today.
Most people are not surprised that there is no ONE thing called cancer. Tumors in all the organs or invasive cells in the blood or bones are referred to as cancer, but start when cells go wrong, whatever the cause. As soon as you are told you cancer, whatever it, the quest begins to find out exactly which cancer it is. With kidney cancer, or its more melodious name, renal cell carcinoma, there seem to be endless variations on what may be called kidney or renal cancer. To treat it requires a very careful analysis of what is really is, starting with the pathology of the tumor when it is biopsied. With kidney cancer that biopsy is usually done after surgery for the tumor. That biopsy will describe the shapes and type of cell in the tumor, which can be mix of types. And then the real work begins.
A recent article in “European Urology” reviewed the mix of HEREDITARY renal cancers, those that arise due to one’s background. More common are the “sporadic” kidney cancer that could arise out of the blue or in response to some environmental toxin. There are ten Heredity Renal Cancers, or HRCs. My goal is to alert the reader to the possibility that his cancer might be one of these. This would affect treatment, and may suggest the need to test family members.
If you have kidney cancer or RCC, you may be familiar with “clear cell” or “papillary” to refine the description of the cells in the tumors. This may not be the whole story, as those HRCs—the hereditary kinds—may manifest a mix of ways, including as clear cell or papillary histology.
The most common HRC is Von Hippel-Lindau (VHL) disease, with both benign or malignant tumors. RCC can be found in a 24-34% of VHL patients, appearing at mean age 39 years (far younger than non-heredity RCC), and often with multiple tumors and in both kidneys. Cysts which appear not to be malignant must be watched–they have the potential to become malignant over time. Generally they are managed based on the size of the largest of these lesions. Clear cell RCC is the one VHL-related subtype.
Hereditary papillary renal carcinoma (HPRC) is rarer, and typically occurs later in life. Papillary tumors are the only phenotype with HPRC, and patients often develop numerous tiny tumors, 1000 or more. These tumors are considered type 1 papillary renal cell carcinoma (pRCC) with a low nuclear grade, monitored with CT scans, and some do metastasize, though this is rare. The MET gene is implicated in the growth of these tumors.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is newly identified as a HRC. Rarely do patients develop RCC, but are susceptible to developing multiple leiomylomas, which are generally benign. When there is early onset of HLRCC, then RCC is found in about 20% of those patients. These tumors can be aggressive, and about 2/3 display a papillary pattern. Such tumors tend to be hyper-vascular.
Birt-Hogg-Dube (BHD) syndrome is quite rare, about 1 in 200,000 people, and thereby likely under diagnosed. This raises the risk of developing kidney tumors, which occurs in 25-35% of BHD patients, and at mean age of 50. These tumors have varying histologic subtypes, generally chromophobe RCC or hybrid variants. And there can be variants in the same tumor or within the kidney. There is a risk of metastases, though rare. The characteristic skin lesions of BHD syndrome are not malignant.
Even more rare is Tuberous Sclerosis Complex (TSC), which can manifest itself in renal lesions, cysts and occasionally, RCC, the latter at a young, average age 28. Neurologic complications can accompany this syndrome.
SDHB-associated paraganglioma/phaeochromoytoma is another heredity condition which may give rise to a mix of renal tumor, including clear cell RCC, chromophobe RCC and oncocytomas, i.e., a mix of histologically different types.
An HRCmay be suspected in patients with a family or individual history of renal tumors, in the instance of both kidney having tumors, or one kidney having multiple tumors or in early-onset renal tumor, i.e., under 50 years of age.
Clinical diagnosis can be further refined by genetic testing, and thorough review by an experienced uropathologist is fundamental to the diagnosis. First consideration would be a VHL analysis and genetic analysis of SDHB and FLCN genes, as warranted. Patients with type 1 papillaryRCC should be considered for MET analysis. The presence of clinical symptoms related to any of the syndromes will guide the gene screening. Testing on family members may well be warranted.
With these cancers, it is possible to have multiple lesions and affect both kidneys. Thus, treatment should preserve renal function and control the risk for metastases. Use of ablation to retain maximum renal function may be preferable to partial nephrectomies, for example.
Though these heredity renal cancers arise in a different manner than the more common sporadic RCC, the study of the molecular pathways provide some insight into new therapies for those patients as well. Thanks always to those researchers who help in this struggle for information, as that is essential to provide treatments.
Why should you care about genomic research? Simple; it could save your life! Want to know EXACTLY which type of cancer you have, and how to choose the best treatment? New hope comes from this research which really examines the nature of the cells that make up your cancer. Pretty important stuff.
Genomic research is bringing improvements to care, and points up the need to be aware of this new knowledge–and if your own doctor is keeping up with that type of data. At a 2012 Conference sponsored by the The Oncology Journal, Dr. Kimrym Rathmell spoke in regard the genomic knowledge that is leading to improved care for kidney cancer patients. Maybe the most critical lecture of late.
Dr. Rathmell begins after introductory remarks; Complete access on YouTube via this link:
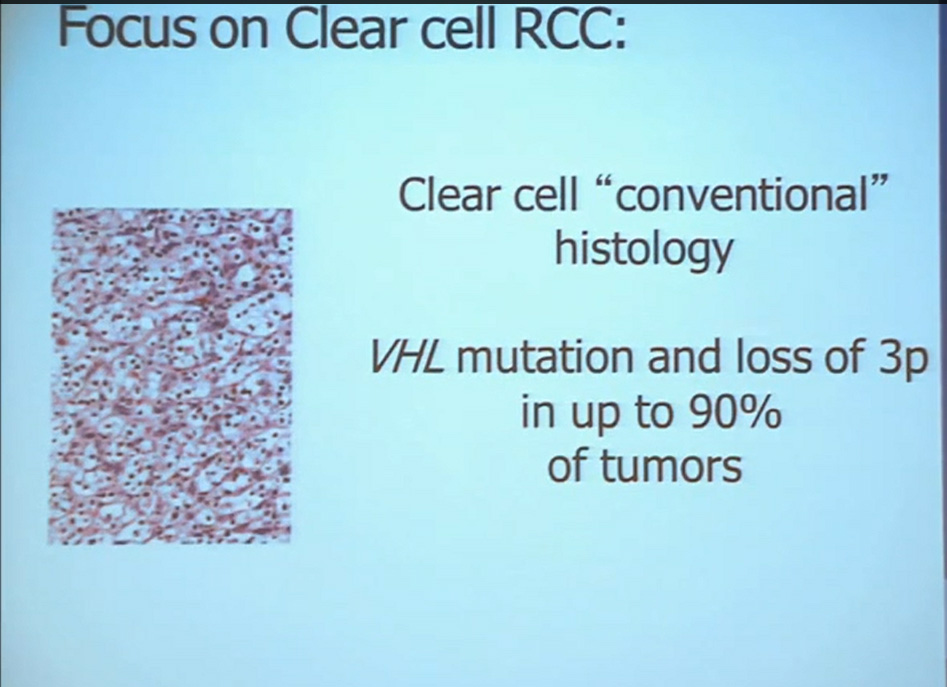
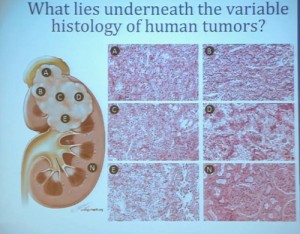
First, kidney cancer, like pancreatic cancer, has been on the rise. This is a somewhat dated slide, dating back to the 70s. We have seen a steady increase in this cancer. Although it was originally characterized as a rare tumor type, it is not really anymore.This talk will focus on one subtype of kidney cancer, that is, clear cell histology renal cell carcinoma. This is a histology slide showing why it is called clear cytoplasm
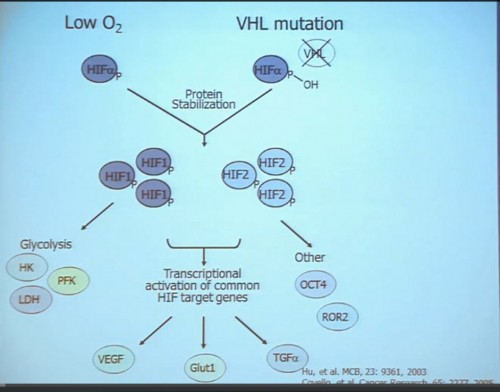
This tumor is characterized by particular mutation. That is the Von Hippel Landau gene, coordinate the loss of 3p, (a chromosome) where VHL is housed. We see these in mutations and loss of 3p which house other tumor suppressors as well, in up to 90% of these tumors. Based on this strong correlation between clear cell renal cell carcinoma and the VHL mutation, a tumor type. is a very distinct paradigm in which VHL loss causes upregulation of hypoxia inducible factors (HIF). These tumors are characterized by loss of high loss of these HIF factors. These are transcription factors that normally allow cells to respond to low levels of oxygen by turning on a repertoire of genes that allow them to bring in new blood vessels, to shift their metabolic properties, to migrate away, to promote survival, and to de-differentiate. That is a perfect storm for kidney cancer, in some respects.
Targeted Agents in Use
Because this cancer has highly nonresponsive to typical chemotherapy, there’s much effort in recent years to develop targeted agents. These targeted agents to date all focus on this well-known pathway in clear cell type renal cell carcinoma. Most of the agents focus far down on this pathway, including that of receptors of VEGF and PDGF. They are tyrosine kinase inhibitors, effective at reducing the tumor angiogenic profile and can be quite effective at reducing the bulk of these diseases. Other drugs similarly target this pathway, for example, targeting features of the tumor that enable HIF to be stabilized such as that in the mTOR pathways. Temsirolimus and Everolimus are approved for use. There are in-developments drugs for targeting MET, which is another mutation that can occur in this cancer, similarly increases HIF levels.
But the reality of treating kidney cancer is that the available drugs that we have do not produce complete responses. We only work in the arena of minimal response and partial response. The extent of response that a patient gets is unpredictable. The duration is also unpredictable and the toxicity is also unpredictable. For drugs we expect them to be effective on average 1 to 2 years, this is chronic therapy, very expensive, and it’s dominated by effects that are substantially detrimental to quality-of-life—fatigue, rash, diarrhea, as well as laboratory abnormalities that indicate damage to the liver or elevations of glucose and cholesterol.
PART I: Clear Cell Renal Cell Carcinoma, Molecular and Genetic Contributions to
INTER–Tumoral Heterogeneity.
With that, I will talk about various molecular probes that we use to understand some of the diversity or the heterogeneity of these tumors across the clear cell renal cell carcinoma spectrum. Before I really dive into clear cell renal cell carcinoma, I need to point out that there are other histologies with this tumor as well. So when we say kidney cancer, we’re talking about a big spectrum. Clear cell renal cell carcinoma, we are talking about those tied to Von Hippel Lindau disease and loss of 3p and it is about 70% of all cases we encounter in cancers of the kidney. But there are also other types. Papillary type renal carcinoma, chromophobe, benign tumor—oncocytoma, a translocation form and some very rare. With these types of tumors we have very little in terms of knowledge of how to treat these patients. Their genetics are highly distinct from clear cell renal cell carcinoma. So someday in the future, we will understand not only how to treat not only our clear cell carcinoma patients, but how to use effective molecular information to target these cancers as well.
Clear cell carcinoma is well know to be molecularly heterogeneous for some time. This is a gene expression profile. We’ve already seen heat maps from several of these other talks, looking at gene expression profiles. And as you can see the gene expression profiles across a large selection of tumors here, suggests there are great areas of variability–at least two and as many as five groups, based upon gene expression purely.
Pattern Recognition Profile to Find Subtypes
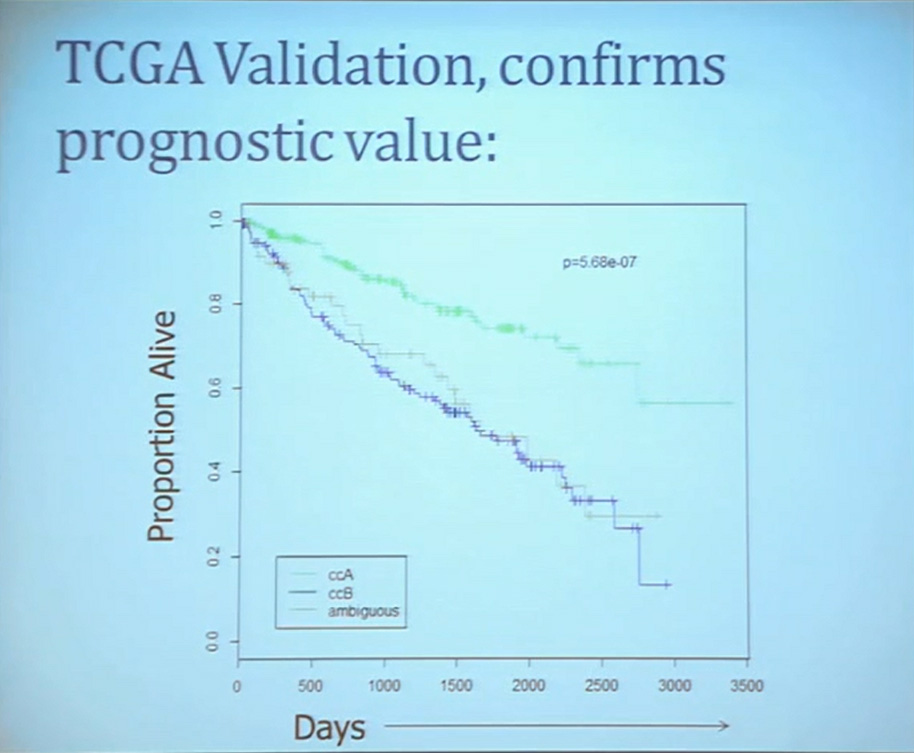
Our group undertook at the time developing a pattern recognition profile which is now fairly routine use. To try to see with a more robust computational strategy what subtypes we could really identify, that we could really pen down and understand with genetic profiles. 8aWe found two. For lack of better knowledge, we are calling clear cell A and clear cell B, ccA and ccB. These are very distinct biologically, and when we look at these tumors in terms of their outcome, they also have significant prognostic relevance–with the ccA tumors in this original cohort having a median survival of 103 months, compared to the 24 months for ccB tumors.
The TCGA which is been discussed here in many of the previous talks is a great source of validation. We assigned these tumors to clear cell A and cc B groups subtypes, validating our previous results with the clear cell A tumors having much better survival profile than those ccB tumors.
This classification scheme, which is based 120-130 gene signatures classified robust subdivisions of clear cell type renal cell can be applied with a small number of genes on individual tumors and is independently associated disease-specific overall survival, making it a valuable prognostic biomarker.
PART II: Rare Variant Groups
We use these profile tools to understand the rare variants. This is still in the clear cell renal cell carcinoma arena, but when we took a very large group of compiled tumors; this was a meta-analysis of 500 tumors, all histologically defined as clear cell type renal cell carcinoma, and we applied our expression pattern recognition algorithm. We asked for two groups and we found two and they correlated with our ccA and ccB, but when we ask for three groups, we can find a small group that now filters out. Now that we have power in numbers to identify what we called Cluster 3. What is in Cluster 3?
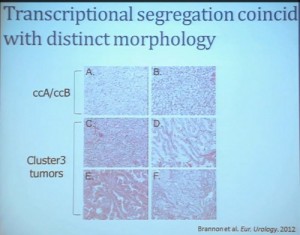
Cluster 3, as we’ve said, is histologically defined as clear cell renal cell carcinomas. But we look to the genetic expression profiles, it’s very different, particularly with regard to metabolic properties. We see upregulation of genes that are involved in mitochondrial regulation and oxidative phosphorylation, suggesting a striking difference in the way these tumors likely regulate metabolism.
In addition, and now these are tumors that we can not go back and genotype for VHL mutation, for loss of chromosome 3p, but the loss VHL regulation leads to characteristic changes in the gene expression profile. So when we use the gene expression changes to predict whether these tumors have an intact VHL or a mutant type VHL. The wild type VHLS signature shown here is shown in purple. You can see that these purple tumors, the wild type VHL tumors all tightly cluster with Cluster 3. These are probably not clear cell renal cell carcinomas although many, pathologists call them that. So we pulled them all out so, asking, “DO they look a little bit different?” My graduate student, who did this work, came right away and said “There’s something funky about these clear cell tumors that we call Cluster 3.”
As you can see, these are clear cell A and a clear cell B tumors, but they all have the clear cytoplasm and really, what we are seeing, is that they are not distinct histologically, although they are very different molecularly. And as I have shown, they have a very different prognostic outcome. The Cluster 3 tumors; although the cells themselves might have clear cell cytoplasm that gave them the clear cell histology designation, they have a very different pattern of organization—with a papillary type of feature. So what we think it would be identified as is a new rare variant of clear cell renal cell carcinoma.
Simultaneously another group of pathologists identified, that the pathologists call clear cell papillary carcinoma. That suggests that we need to take a very great care as we treat these patients. What we have is clear cell type renal cell carcinoma, most of which are VHL-mutated, and we do have clear cell A and clear cell B. These are the tumors we should be treating with the drugs with identified, based on the effect of the pathway that is activated by loss of VHL. But clear cell papillary renal cell carcinomas probably won’t react very well, as they are VHL-wild type. Just like papillary renal cell carcinomas don’t react well either.
To summarize this section, clear cell renal carcinoma can be separated into ccA and ccB groups, based on transcript profiling, but further clustering can identify highly biologically dissimilar subtypes within the clear cell group, and that subtyping can convey a biological distinction which is a valuable tool for prognostic evaluation, and a likely cause of poor responses to some therapy.
Part III; Using Clinical Trials to Understand Biological Relationships to Response to Therapy
As my title indicated, we also refer to clinical trials to help us understand renal cell carcinoma a bit more. A clinical trial we completed some years ago, LCCC0603, was in neoadjuvant trial that looked at the treatment of renal tumors with sorafenib. Patients were identified as having renal tumor and underwent CT scans for basic size, description and PET scan, and then were treated with sorafenib. It is the first generation VEGF receptor tyrosine kinase inhibitor for 4 to 8 weeks, and then underwent post treatment CT scan, PET scan and a nephrectomy. We are going to look at radiographic indicators of response, rather than molecular indicators.
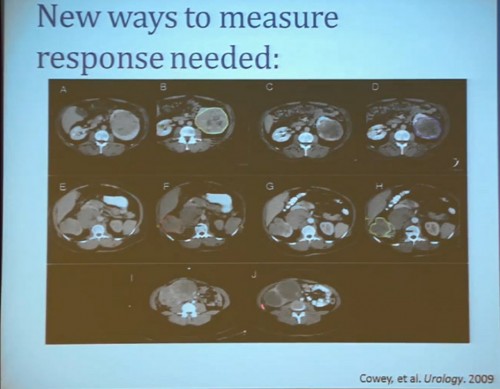
Waterfall Graph with Response at 30% as goal
First standard RECIST criteria do show that we do see partial responses. Again, there were no complete responses. Many had subpartial or minimal or some partial responses. Their tumors shrank, but most did not meet the standard criteria of 30% decrease in one longest size. Some tumors actually grew.
Now what we realized as we looked at these tumors, is that we probably need new ways to describe response. The standard RECIST criterion response is just based on longest diameter and measuring this in comparison, after treatment..
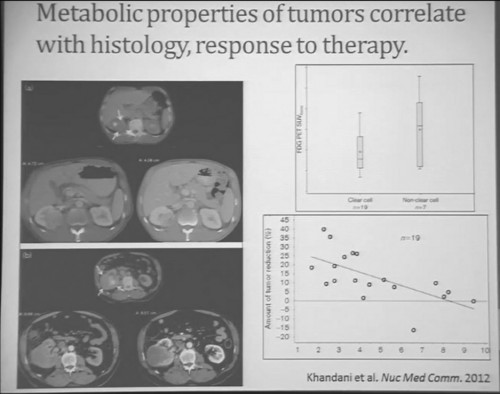
I will use this patient is an example. (References I and J, in the lowest row). Here is a pretreatment; we have a very large renal tumor. Post treatment, the tumor was still large, but it measures slightly larger than it had been before. But if you look at this tumor, it is very different. The central area of this tumor is now very dark, indicating necrosis—is what we think. But we took these tumors out, we could confirm that these dark areas were indeed necrotic. So we developed a new way to try to quantify the area of the tumor that is actually killed in response with this treatment.
Similarly we were doing PET scans on these patients, and we were doing this because we’re trying to understand how the metabolic properties of these tumors might indicate how these tumors were likely to respond to this treatment. We see, and have known, that are some tumors which are very dim on FTG PET. This is a tumor; (Smallest of upper images) you can see that here is very visible on the PET scan. It doesn’t take up any FTG. So this tumor has the metabolic profile that is not dependent upon uptake of glucose. Others; this tumor (Smallest of lower picture), for example, have regional areas that are can be very high in terms of FTG uptake. When we looked at these tumors, we discovered first that non-clear histology tumors were much more likely to have high levels of FTG uptake. So, metabolically active tumors more likely in the (correction) non-clear cell group, probably the papillary, the chromophobes and the papillary clear cell types, than the clear cell group. Secondly. we discovered that the correlation between FTG uptake and response looked somewhat different than we might have expected. We might have expected the most metabolically active tumors would be those that would response better to anti-angiogenic agent. But the opposite was true. The best are those that had very low levels of uptake FTG uptake. We are still trying to understand exactly what that means. Certainly that means that those clear cell tumors are the ones more like to respond, activity, what we have known, but those are the ones with the lower level FTG activity. But we continue to try the metabolic properties of the tumors that make theme different more likely to respond.
That leads to our next clinical trial. This is now ongoing. This is LCCC1028. It is a neoadjuvent clinical trial using the newest generation—well, they are coming out so fast that it’s the not the newest, but the next to newest VEGF receptor tyrosine kinase inhibitor. They are now getting PET scans and a biopsy to confirm in fact that they would be clear cell renal cell carcinoma, and also to allow us to do molecular studies that directly measure their metabolic activity and other effects. They’re being treated for eight weeks with another CT scan, undergoing nephrectomy. We will then be able to look at clear cell variant histology. They will all be clear cell going in, but there may be variants included–as well as looking at their VHL mutation and their other mutational status, their transcript profile, in particular the clear cell A and clear cell B group and other protein expression signatures.
This, of course, is known for all tumors, but if you sample in multiple different places, the histology will look different and in effect, the grade can look different depending on where you are sample.
What does that mean molecularly? Well, a group at the Sanger Center published on a small number of tumors. When they sequenced these tumors, they found while there are some mutations that are ubiquitous, meaning the mutation is found in all samples across the primary tumor and the metastatic tumors, that there are mutations that are private. There are mutations that are common only among the primary tumors and there are mutations that are common only in the metastases, and there are a lot mutations that are unique to the individual sample. This makes a whole new level of complication as we moved toward personalized therapy, in particular therapy that is based on biopsy metrics.
This group also looked at our clear cell A and B subtypes. And what they saw, when they looked at six samples from the primary tumors, was that in five of those samples, the gene signature indicated that these would be clear cell B type tumors. So depending on your glass half-full/glass half empty: The glass half full version of this, that five out of six times, they would pick that the patient would have poor outcomes. This patient has metastatic disease, so it fact, that is true. The glass half empty would be that one out of six times, he would pick wrong. This patient would have been indicated to have clear cell type A tumor, and you might have predicted that this patient would do well, when in fact that would be wrong. So what helps us understand the limitation of this test. It also gives us the opportunity to understand a little bit more about these tumors.
(Footnote reads: BRIC Funded Grants LCCC1213) So for the future, a trial (LCCC1213) that we have really just initiated is uniting some of these imaging observations we have made with genetics.
We are taking in patients. This is patient number one. Patient number two just has his MRI last week. and doing an MR in coordination with the PET scan so we can get the detailed look at these patients’ tissue perfusions, vascularity and the density of these tumors, as well as regional areas at the FTG uptake and sample according to the map that is created by the imaging, as well as samples that are collected, based on what we see grossly in the tumor. Here you can see a sample that we collected from a tumor region that is highly distinct from the mostly more pale yellow regions of tumor.
This is just begun, so I cannot tell how well were going to get to correlate the gene expression and genetic underpinnings, and what we see in the tumor and what we see in the MR / PET. But it will help us to move forward.
To summarize are multiple ways for RCC to diverge. The subsets can enrich tumor sets for clinical and genetic features, and a multiplatform approach that with genetics, molecular biology and imaging techniques will give us man ways to tackle a surprisingly very heterogeneous disease.
Probably the most talked about studies at the June 2013 ASCO in Chicago swirled around the newly released trials about PD-1 (Programmed Death-1) and similar. “What the heck is this all about, and why should I care?”, you may be asking. This is important as it shifts attention back to the original immune therapies that kept me alive (and many others) when there was nothing else to offer any hope in the olden days of the 1990s.
Though a number of targeted therapies have since emerged, and you have been hearing about them, there is new interest in the earlier and modestly successful immune therapies. I confess a fondness for anything described as an immune therapy, as I am alive–which I would NOT have been–without the first FDA approved agent against kidney cancer, high dose interleukin 2, brand name Proleukin.
The first new agents in the 2005+, the anti-angiogenesis drugs (not easy to pronounce,much less to understand) brought new hope to metastatic RCC patients. With the late diagnoses of many kidney cancer patients, we desperately needed hope. For about 14 years, HD Il2 (high dose interleukin 2) was the only game in town. Nothing else existed, so any patient lucky enough to hear about it, and not actively discouraged by the very “realistic” doctors, probably considered it. This is a hospital-based agent which revs up the immune system, so the immune system would go after the metastatic disease, at least for some number of months or years. That is why I am alive. Nothing else was offered and nothing else was available. Nine years!
Since that time the anti-angiogenesis drugs–translation: those that fight against (anti..) the creation (genesis) of angio (think blood vessels) drugs were approved. Think Sutent (sunitinib) and Avastin (bevacizumab) and others,. They have been the weapon of choice for oncologists and patients. Why is that? Though these drugs rarely offer more than some pushing back of the drug, decidedly welcome, they have rarely given more than some slowing or relief from the metastases, and always with some side effects.
For us old-timers, who faced only death, whether by efficient means or by surgical cut-and-pastes as holding actions, this was a tremendous breakthrough. For these options, even if only holding actions versus mop-up operations, we were grateful. We who had nothing previously were slightly scornful of those who complained about the side effects, as we were grateful to be around to have such effects. The Stage IV sufferer in 2004 knew too well that Stage V was a damn unpleasant journey and without a return ticket.
Now we have become greedy again, wanting more than the “stability” or “some shrinkage”, which I applaud. We want success and life, not holding actions. High dose interleukin 2 continues to offer that success to patients, but no one can predict which patients and which conditions that might be likely.
Bless those researchers who continued to wonder why kidney cancer and melanoma (and maybe non-small cell lung cancer) respond to some sorts of immune therapies, and what happens to make that happen and not happen. Those fine people have come to understand that the immune response which is revved up by HD IL2 has a complex set of “calls and responses” which either let loose the dogs of war/and immune responses, or fail to do so.
Most people understand that the body tries to protect itself from assaults, whether by poison ivy, bronchitis or cancer. The immune system responds, gives the body aches, pains, fevers, chills, etc. Think of flu and how rotten you can feel. Think also of old diseases like Black Death which stimulated the immune system so violently that it was the strong and healthy with good immune systems that succumbed to the immune response, dying with lungs filled with immune reactions and fluids. Only those with weakened systems and slower responses managed to live through the symptoms.
Obviously the body’s immune response needs a moderation, and not one that saves the village by killing it. All of this leads to an explanation of the newest immune therapies, now in clinical trials in RCC. You may hear about PD-1 and CTLA-1 trials, and how they may be helpful in kidney cancer, melanoma, and now in non-small cell lung cancer.
To understand all of this, it is helpful to think of a system of checks and balances, perhaps in an electrical or computer communication system. Just as every electrical impulse might be appropriate, it could also do damage by overwhelming the system, burning it out, or failing to meet the requirements to be able to answer a “send me” signal. So it is with the latest research on immune therapies with the PD or Programmed Death agents.
Keep in mind that the immune response is typically used against infections, and not generally against cancer. I assume that we cavemen needed more protection evolution-wise against infections and less so against the ravages of cancer, which seems to result from getting older and getting overwhelmed by changes/mutations in the body. (Not always, I know, but that is a discussion for later.)
T cells are supposed to react to a number of infections and such, and trigger an immune response to fight back against the “aliens”, such as cancer and ragweed. Just right amount of fight, and the body recovers. Too little, and you have the devastation of the Black Death, an over-the-top immune response.
The immune system is supposed to respond to handle the natural threats to the body, but not over react and set the system on fire metaphorically. It had built in checks in balances, as do normal cells. We see this everyday when our normal growing hair cells decide to stop growing and the hair fall out. Chemo patients struggle with the balance of killing all cells, with the faster-growing cancer cells being killed off first.
New agents have been developed that interfere with a signaling system that puts the T cells, the protective/fighter cells into action against cancer cell. This naturally happens, but the nasty cancer cells try to evade that process by interfering with that process. Not letting the body protect itself, by disguising itself as the evil twin, equally eager to live, as the good twin, the healthy cell, cancer interferes with Programmed Death. More next time…
And as Mae West said, “I like a man who takes his time.”…
If anyone can find a cancer event exciting, it is the ASCO annual meeting, at which thousands of oncologists present their studies and learn from one another. In the kidney cancer world, the “buzz” has been about “programmed death”–for the cancer cells, which we all prefer over that of our healthy cells.
This is a transcribed lecture, fairly short, to explain the immune therapies that may add to those weapons against RCC. Dr. Wolchok clarifies how this happens, with the agents interfering with the “Shields Up!” commands that protect the cell from what should be their programmed deaths. With appreciation to all researchers.
Peggy–Nine years of life free of cancer, thanks to the original immune therapy, HD IL2
ASCO Daily News June 6, 2013
Dr. Jedd D. Wolchok, MD PhD of Sloan Kettering Memorial Cancer Center
We now recognize that there are several pathways that constrain T Cells and the immune system from achieving a state of full and persistent activation.
One of those pathways is called the CTLA4 pathway and there was a medicine approved call Ipilumimab last year, that blocks CTLA4 for treatment of melanoma. There is another pathway, the PD 1 pathway which usually constrains the immune system in a different way. And PD 1 is also present on T cells in the immune system and it binds to a family of ligands, one of which is known as PD-L1 or PDL ligand 1.
In the ever-expanding a list of ways in which cancers try to evade the immune system, cancers have learned to express this PDL-1 ligand on their surface. By doing so, they actually cause the death of T cells that come close enough to them or PDL-1 to bind to PD-1 on the T cell surface. This ability of cancer cells to express PD L1 on their surface allows them to defend themselves against T cells that the body is trying to educate to see the cancer and to defend itself.
So the PD-1 pathway is active across multiple disease sites. Some of the earliest work done with anti-bodies that block the PD -1 pathway used an anti-body called Nivolumab. Nivolumab was used in a phase 1 clinical trial that was presented at ASCO last year, and additional data will be presented this year.
The data last year showed that the antibody Nivolumab blocks PD -1, and that it can cause regressions in melanoma, in kidney cancer and importantly, really, non-small cell lung cancer. It is not traditionally recognized that as a cancer amenable to immune intervention. But when you people talk about cancers that responsive to immunotherapy, melanoma and renal cell carcinoma come to mind.
Now I think with the data generated first with Nivolumab and now with another PD-1 blocking antibody called Lambrolizumab(MK-3475), these anti-bodies which block the PD -1 pathway are demonstrating activity outside the “usual suspects”, melanoma and kidney cancer.
This ability to affect multiple different cancer types is important, as it shows that immunotherapy is not a treatment for one particular kind of cancer,. It is a treatment that primarily targets the patient, Then it is the patient’s immune system that goes out and treats the cancer.
One of the most important characteristics of immunotherapy is its ability to induce durable and certainly there are many different types of anticancer medications which can cause a tumor to reduce in size, but the challenge has been and continues to be, how to get disease to regress and stay regressed. Tumors, because of their genetic instability, can find pathways to become resistant to these interventions such as chemotherapy or targeted pathway inhibitions.
Immunotherapy really falls into a different category. Again, it is not targeting the tumor itself. It is targeting the patient’s immune system. It is causing the patient’s immune system to respond to certain parts of the tumor cells that the immune system finds interesting and then to control.
We know that the immune system has the ability to remember, through to a population of cells called memory cells. Because the immune system is really a dynamic organ that cannot only sculpt itself around changes in the cancer, but also can remember what it has been exposed to in the past, we believe that durability is in fact a hallmark of response to immunotherapy. The first medicine to show us this in a meaningful way was a drug called interleukin two which was really actually developed now over 25 years ago and has led to the cure of some patients with melanoma and kidney cancer. That medicine is a hormone that causes the growth and differentiation of T-cells and patients who have a complete response to IL-2 and remain in complete response for at least two years don’t ever seem to recur with 10 or more years of follow-up.
We definitely need to learn more about the PD-1 pathway we specifically need to know whether it’s absolutely required for a tumor cell to express PDL-1 that on that surface to benefit from PD-1 or PD L-1blockade. It would be ideal, in fact, if a predictive biomarker that could identify the precise patient population who would benefit from that intervention. However, I think that is not going to be a simple as predictive biomarkers been for some of the targeted therapies, where it is mutation-present or –absent. Here PDL-1 is not just important when it was expressed on the tumors, but PDL-1 plays a role by its presence on antigen-presenting cells. So, in its normal physiologic role, the PD -1 pathway actually involves interactions between T cells and antigen presenting cells and by blocking cells–even if the cells doesn’t express PDL-1–one could imagine that a patient could benefit, perhaps not as likely as if the tumor expressed PD L1.
I love this study, as it really symbolizes the tremendous change that has occurred in kidney cancer treatment these last 6 years. It is remarkable that the 124 patients are described as having already received first- line treatment, and were now in their second-line. These same metastatic patients might have received neither just six years ago.
The study was really not to compare the response to the treatments received, whether Sutent (sunitinib), Nexavar (sorafanib), or placebo. (Why any party chose or was chosen to receive a placebo is another, darker question.) That the median follow-up was 80 months is a triumph by itself. This is in contrast to the clinical trials that often show just a few months extra time which we and politicians can focus on, when the reality of much longer responses is clearly shown here. Of course, these longer survival times came from those trials which showed those few months–and this shows the reality of many more months and years of life!
Metastatic tumor burden(TB) was measured, based on the size of the sum of the longest unidimensional diameter of each targeted lesion. The additional increase of 1 cm (about 3/8”) was significant in predicting response to the medications. Siimply, adding the one-direction measure of the lesions and comparing them showed that more tumor was a bigger problem.
One can also assume that to remove as much tumor as possible may be helpful in maximizing the benefit of the meds given, although this study does not address the actual types and locations of the mets, nor indicate why no other therapies, surgery or ablation, were used. With 124 patients this would represent a mix of individual experiences, more like the typical patient group.
What does “median follow up of 80 months” really mean? A median is not an average, but a measure of the time point at which ½ of the population studied had follow up less than 80 months and ½ had follow up for more than 80 months. Since this is considered a long time in clinical trials and becomes more of a longitudinal study, we may never know the average length of time that these patients had either PFS (Progression Free Survival—time until the mets began to grow again) or OS (Overall Survival). In any case, we are aware that following this second-line of treatment, there are still more therapies and interventions which may be available. And even more options are up for FDA approval as I write.
All these options and the greater success of each muddies the study waters, but clarifies the hopes of those with metastatic RCC, or are at risk. This study proves that tumor burden (TB) is a disadvantage. Most patients have naturally assume that more cancer is worse for you than less cancer—who knew? But this gives weight to the notion that the removal of some tumors, if not all, can be beneficial used with targeted therapies. In the past, some oncologists have discouraged additional surgery in the light of metastases, with the implicit message, “It’s too late, and won’t help you anyway.” Not the doctor for me.
The story is quite different right now, but patients may need to tell this to their doctors–in the language that the doctor speaks. Certainly, there was a time at which doing more surgery for mRCC patients added little, if anything, to survival and probably even less to the quality of life. That no longer is the case, and those older studies no longer have meaning. While each patient must be treated as an individual, in light of all the variables that impact his health, there is increased optimism for the metastatic patient. Aggressive and early treatment can no doubt extend life and make it worth living.
Halfway through my high dose interleukin 2 (Proleukin) treatments, having completed weeks 1 and 2 and into the second rest period, I got the second most important scan of my life. The most important CT scan changed my life, with its image of a tumor larger than a baseball, and countless tiny specs of white death in my lungs.
This new scan in August of 2004 would let me return to the hospital for more of the immune stimulant, Proleukin, which revved up my internal immune system to the max. My system was then to recognize and fight off the residual kidney cancer that had settled visibly in my lungs and any other new sites, as yet unseen. The new vocabulary of cancer forces me to say that while hundreds of tiny lung mets/lesions/nodules–no wonder we patients get confused–were not just there, but ” visible”. Implicit was the message that many hundreds more were invisible, certainly alive and thriving, just not yet “imaged”.
The tools of imaging cancer are varied and unequal, some more effective for some cancers, and yet inadequate for others. First of all, the tumors are measured in centimeters and millimeters, and that language shift alone makes them even more inaccessible to Americans. Just how big is 9 millimeters anyway? And a centimeter seems a pretty vague measurement after years of holding thumb and forefinger together to how 1/2 inch with decent precision. Is a change from7mm to 9mm cause for alarm?
Plus the panic of hearing “cancer” drives any math computation out of one’s head. The only math question that can be asked and then not understood is, “How long have I got, doctor?”. And then there is the matter of what can be seen with which instrument. When doctors tell smokers that their x rays are clear, naive civilians translate that to mean their lungs are free of cancer. A rough interpretation might be more like, “Your tumors–if you have them–aren’t big enough to be captured by this 100 year old device. Come back when we can see something/you are pretty much past help.” Notice that I provide the translation here.
The x ray is more like using a child’s microscope to look at something; great for noticing crud on your pet’s hair, but not quite like those grownup electron microscopes which can see cells. Note to kidney cancer patients–don’t even bother.
Another imaging device is a PET scan, which measures the activity of cells, and which I understand a quick snap shot of liveliness of the cell, and it manifestation, the tumor. Is it chowing down on the body’s nutrients faster than the orderly cells? That activity will “light up” in a PET scan, so the lazier cancer cells might be overlooked. Not to effective in kidney cancer, whose tumors are often slow-growing. That is also possibly why kidney cancer can establish itself so thoroughly in so many patients, not noticed until a broken rib or vague back ache or a non-existent ulcer finally results in a CT scan. Note to lots of kidney cancer patients–don’t even bother with a PET scan.
So a CT scan, with contrast to enhance the vague and ghosty images is the way to go for most RCC patients. A blood test to see if the single kidney can handle the assault of the imaging fluid precedes the test, and then the patient settles onto a big padded tray which is drawn slowly into a large doughnut-shape machine that somehow can see into the patients insides. I’m not even trying to explain that.
That exam is not painful, not pleasant, and not anything you can study for; you simply submit, as patients are supposed to do, and then the impatience begins. Most patients must wait to get their results from the doctor, and this is the longest wait of one’s life. Am I dying more efficiently than before? Are the cells multiplying more quickly? How long have I got, doctor?
But I had learned the secret, which I now announce to all. You are ENTITLED to your own reports, and with a bit of research as to when they get read, you can go get them. Apparently for many imaging centers, this comes as a surprise, but we are all grownups and can explain that. Sometimes your doctor will need to hear that too. I have always just called ahead to the “Medical Records” department and asked for the report to be ready, as I have no patience…again.
But in August 2004, I had to be outside before I could read it. To read a report of impending death inside a hospital with its metal window frames and linoleum floors and sad, bent people waiting their turns is too harsh a setting. Immediately stepping outside, I could rip open the envelope, and read that my lung tumors were shrinking, even those big ones. The 13mm lesion was now 8x9mm, the 8x7mm lesion was just 4x4mm. And the countless other unmeasured one? They were likely shrinking, too. And 9mm is .354 inches, which I still can’t measure between my thumb and forefinger, but the CT scan could, and that was good enough for me.
Back to the hospital, a much easier trip than before, to get two more week-long sessions of HD IL2. The mets were shrinking and I was getting more ammo against them. The cloak of invisibility was pierced and my immune system was working again.
My first week at home following the CT scan was to be a recovery week, following five days in the hospital. When I had anticipated that first weekend, post Proleukin/High Dose InterLeukin 2 treatment, my plan was to go to a local kidney cancer meeting. I would be a bit soggy, post “flu”, but not contagious, just recovering from the immune stimulant. I planned to sit around, smile bravely, and look like those heroines in the old days of consumption.
As a measure of my mental competence, it was several weeks later before I realized I had missed the meeting, and in fact, had missed most of the week, and a good portion of the recovery week as well. My response to the Proleukin was such that I did not receive but 9 of maximum 14 doses and had to spend an extra day in the hospital to recover. The last thing I do remember of that week at UCLA was a doctor walking me down the hall, and seeing a sign overhead. At least I could read, and knew I was in the hospital, and then somehow I was home.
Flashes of memory come to me of those first few days, retching in the toilet, and then being sat in the shower on a tiny plastic stool, and being scrubbed by my daughter, a miracle of an experience. And more odd oblivion. At this time, my mother-in-law was headed deeper into an oblivion of Alzheimer’s, and I joined her. One WEDNESDAY morning, I was reading the paper v e r y s l o w l y…not my usual style, and I noted that the LA Times used Tuesday’s date. Obviously this was major mistake, which rather incensed me, and tried to interest my husband in this crisis. “What day is it?” He also thought it was Tuesday! For that matter so did the local paper, and the Wall Street Journal!
Naturally, I had misread or misheard this, so checked again, and asked again, and asked again, and read again. Even the damn computer was in on this mistake, but I waited patiently for corrections to occur, and still they–now the entire world–claimed it was Tuesday. And the family was getting a little odd in their responses to me. “Still Tuesday, Mom…”.
Not once all that Wednesday nor the Wednesday that followed it did I ever think for a second that I might have been mistaken. My testing, my logic, my checking and rechecking all these reliable resources did not change my mind, but it did cause me to wonder why everyone else was wrong. I figured the nice thing to do was to wait until they got it right.
It took me several months and a number of similar events to realize that this was a tiny gift from HD IL2–and insight into craziness, or whatever word describes the inability to accept fact in the face of facts from trusted and even beloved sources. No wonder poor Nana could get so angry at all of us, and no wonder that telling her something in a logical manner was futile. Her brain had been compromised by Alzheimer’s and mine by the medication that was trying to save the rest of me.
So was it working? Brain issues aside, I felt fine, or so I told my oddly polite and amused family. Taking a plate to the sink proved to me that I was doing all my household duties. Writing a 25 word email in 30 minutes proved my computer skills were intact. But what about those precious lungs and the icy white granules of tumor dividing relentlessly?
Without proof that the IL2 treatment was slowing down the growth, I would not have been permitted back into UCLA. To push the immune system into the kind of response that causes it to seek and destroy the cancer cells so well-settled into my lung, not only the visible ones, but their countless and invisible spores, is dangerous. No doctor wants to make a patient sick without hope that this synthetically induced sickness would drive out the virulent and relentless cancer cells, so it had to be shown to be worth the risk.
As I really became aware that I had missed one week and more in my life due to the treatment, and that I had no control of memory over what had happened in the hospital, I realized how hard it would be to readmit myself to the hospital. Excuse after excuse–all good ones, of course–delayed my walk into the lobby, accompanied for the first time by fear.
Years earlier, as my father lay dying in our family room, he told me that he was afraid to go to sleep, for feared he would wake up dead. We grinned wryly, and promised not to let that happen, though it did–everything but the waking up. I was raising my hand and volunteering to do just that, walking back in through the gray and damp parking entrance to UCLA.
Coming out the second week with equal blank spaces and some low blood pressure “events”, I was still alive, and could plan for my CT scan. Back to the same place where they had first found and failed to tell me of the lung mets, and let them try again to “image” them. (Is image now officially a verb?) One thing remained in my brain, and that was the knowledge of how to get the report from the CT scan done two weeks plus into my rest period after week two.
My son drove me to the scan center, as I was oddly not thought to be capable of going there myself, such a smart family, and parked as I rushed to collect the report. It was at the desk, as promised, and I tore open the envelope, and pulled up the last few sentences of the report into view. “Significant decrease in size of multiple pulmonary nodules!”
And back for more Proleukin. And a cake that I ordered for myself, which read (Charlotte’s Web alert) “Zuckerman’s Famous Peg…Amazing.”
Dr. Suzanne Topalian, professor of surgery and oncology at Johns Hopkins Sydney Kimmel Cancer Center presented a paper at a recent ASCO meeting which caused quite a stir in the kidney cancer community. Her short talk is in the link below, with the transcription to follow.
What’s the good news about being anti? The complex interplay of our immune system and the manner by which cancer escapes its notice is a challenge to the researchers, but this trial shows that there are many ways to interrupt the growth of cancer cells. This trial and another mentioned offer new hope to patients who have already exhausted earlier options.
Not only did this trial show that this drug could provide relief to some patients with kidney cancer, lung cancer and melanoma, the presence of this anti-body may serve as a biomarker, and may predict which patients might respond to the drug treatment. Another step forward and more hope for all of us.
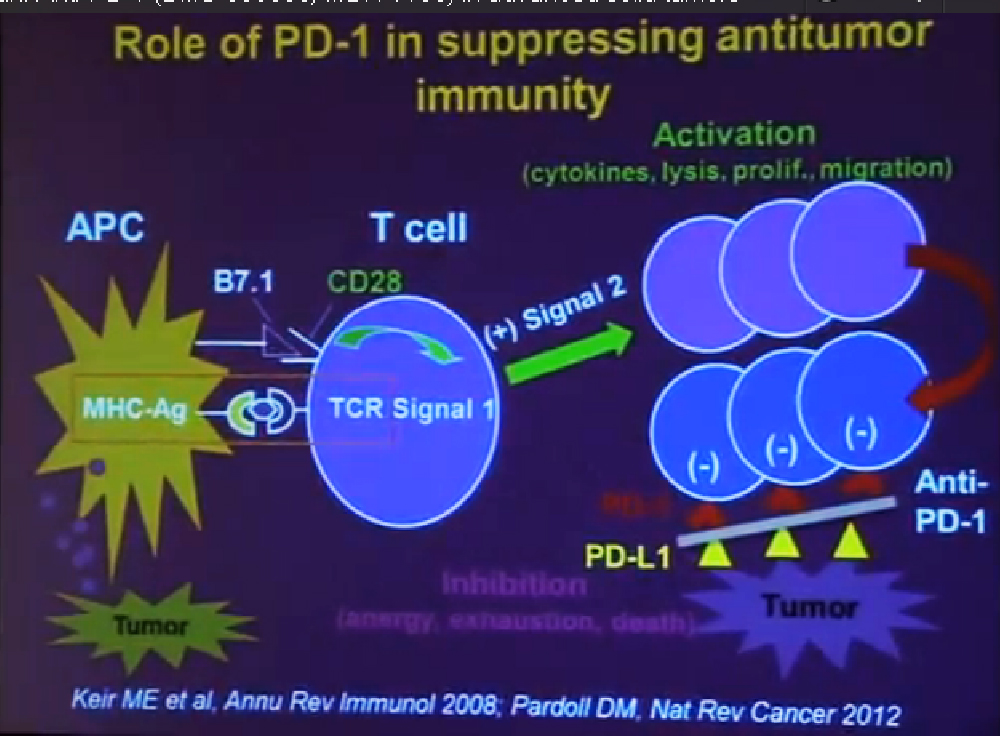
Today I would like to describe do the clinical activity the safety and potential biomarker of clinical response to the drug PD-1, which is an anti-body therapy. PD-1 or Programmed Death-1 is a molecule that is expressed on the surface of activated immune cells it plays a very important role in suppressing the tumor by suppressing antitumor immunity.
In order to understand how anti-PD one works you need to understand a little bit how the immune system works, and how it can fight cancer. T-cells are a central cell type in the immune system that fight cancer. T cell function is regulated by two different signals. Signal one is a specificity signal, whereby the T cell recognizes its target and here we are talking about the targets being components of tumor Cell., but then you need a second signal to tell, the T Cells what to do, a regulatory signals. That signal can be either positive or negative.
If the signal is positive or stimulatory, t he T-cells become activated. They secrete cytokines. They can kill tumor cells. They proliferate; they percolate throughout the body, seeking out and destroying tumor cells. All of that is what we want to see.
But after activation, T-cells naturally begin to express the molecule PD-1 on their surface. This is will turn the T-cells off. If they encounter the partner molecule PD-L1 or PD ligand 1, tumors cells can express PD-L1. So the interaction between these two molecules becomes a protective shield, that shields the molecule from immune attack. Even if the T-cell can recognize the tumor and they can get to the tumor, once they get there and they are expressing PD-1, if the tumor is expressing PD-L 1, the T-cells will be turned off. The anti-PD-1 antibody is a blocking antibody to PD-1. It interrupts this interaction and functions to rescue exhausted T-cells and to enhance anti- tumor immunity.
The phase 1 trial of anti-PD-1 that I’m describing today is a multi-dose regimen in which something is given the outpatient in the outpatient clinic once every two weeks. Patients were treated for a cycle of four treatments over eight weeks. At the end of which, they were restaged. Patients were eligible for these trials if they had advanced metastatic melanoma, kidney cancer, lung cancer, prostate cancer, colorectal cancer with progressive disease after having had at least one prior systemic therapy.
But they were allowed to have up to five of the therapies. Generally these patients who came on this trial had good performance tab status. But they were heavily pre-treated. Approximately half of them had at least three prior therapies be before they came onto the trial.
After the first cycle of treatment if patients had rapid continuation of disease or clinical deterioration, they went off study. If they had unacceptable side effects, the patient remained on study. They did not receive any more drug, but they continued under observation. If the patient demonstrated tumor regression or stable disease or even if they had some progressive disease, but were clinically stable, we continued to treat those patients until we saw confirmed Complete response, worsening or progressive disease or unacceptable toxicity. We could treat patients on this trial continuously for two years. After, they went into a follow up phase.
Here I’m showing you the drug-related adverse events are side effects that occurred in at least 5% of 296 patients which was the total patient population on this trial You can see that serious side effects were encountered in 14% of the patients. The most common side effects are listed here (fatigue, rash diarrhea, pruritis, etc.) There other side effects that are not listed here because they occurred less frequently. Many of the side effects were consisted with the side effects with over immune related causality as you might expected if you release the brakes on immune responses. As we are seeing anti tumor responses, you might also see immune-related sided effects.
We did see three treatment related deaths on this study. This was in 1% of the patient population due to pneumonitis, or lung inflammation which we’ve believe has an immune-related etiology. Over the course of time we developed better ways to identify people who are at risk for the side effect and also better ways to detect it early on and to treat it aggressively.
Also note that only 5% of all patients treated on this trial had to discontinue treatment, due to related side effects so in general the treatment was well tolerated in an outpatient setting, and in general the side effects were manageable.
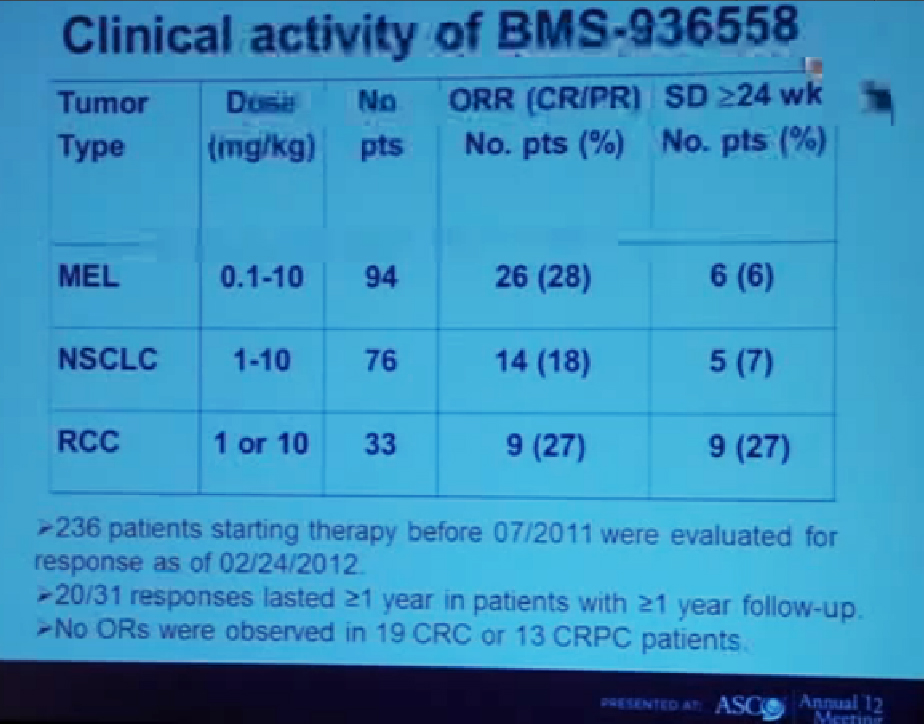
This is showing the clinical activity of anti-PD-1 antibody in three different types of cancer across a wide range of doses. (Showing doses (mg/kg) of 0.1-10 for melanoma, 1-10 for lung cancer, and 1 or 10 in RCC). The largest number of patients in this treatment population of 236 patients who had at least six months of follow-up were 94 with melanoma. We had 26 patients (28%) who had objective responses. An objective response means either a complete response or a significant partial regression of cancer.
We also saw stable disease that lasted at least six months in another 6% of patients. Among lung cancer patients we saw patients with squamous as well as non-squamous subtypes we saw a CR plus PR of 18%, and with a patient population of 76 and again 6% with stable disease, with another group of patients with stable disease (referencing 7% of lung cancer patients.)
Finally in kidney cancer (33 patients), 27% had a response rate and 27% who had prolonged stable disease. There were 31 patients on this trial who had a response that occurred at least one year ago and among those 31 patients, two thirds of them had a response that persisted for more than one year. One of the remarkable features about this therapy is that it can induce very durable responses in otherwise treatment-refractory patients with advanced disease. We did not reserve any objective responses in 19 colon cancer patients or 13 prostate cancer patients.
Finally I’d like to draw your attention to a possible molecular marker that would allow us to predict which patients are most likely to respond to therapy.
In a subset of 42 patients on this trial, we examined pre-treatment tumor biopsies for presence of PD-L1—and again this is the partner molecule to the PD-1 that is expressed on tumor cells. What we found was a correlation between the expression of PDL-1 on tumor cells and here I am showing you the pre-treatment staining biopsies.
I am showing you with its ringed expression an example of melanoma, kidney and cancer in a sample of lung cancer. When we saw this kind of expression in that group of patients we had a 36% objective response rate. If we did not see that expression on the surface of tumor cells we did not had no responders. I would stress that these are very preliminary data but give us an important lead for further investigations and potential biomarker development.
In conclusion anti-PD-1 antibody–BMS 936558– can be administered safely in an outpatient setting for heavily pretreated patients with durable clinical benefit for patients with lung cancer, melanoma and kidney cancer.
These results will be released tomorrow, as you know in the New England Journal of Medicine, which is under embargo until early tomorrow morning. At the same time in the New England Journal, there’s a companion paper with the blocking antibody against PDL-1. The lead author of that paper here is Dr. Julie Braemar of Johns Hopkins and shall be available to answer questions at the end of session.
We found responses also in melanoma and lung cancer and kidney cancer with a blocking antibody against PD-L1, so we feel these two studies are in a sense bookends to point up the point the importance of the PD-1 pathway in cancer therapy across multiple histologies.
The preliminary data correlating PDL-1 expression in pretreatment tumor biopsies with outcomes needs to be further explored and that’s an area of active investigation. Finally controlled clinical registration trials of this drug with patients with the three types of cancer that seem to respond are planned. Thank you for your attention.



















 QED
QED