With the headline, “Nivolumab Shows a Substantial Objective Response Rate in Refractory Non-Clear-Cell Renal Cell Carcinoma”, the article should be welcome to all of those in the in the non clear cell RCC world. Clear cell is the most common, the garden variety of renal cell carcinoma. This is welcome news, as the non clear cell patients get very little attention from the research world. Though the patient with nccRCC might interpret this as, “Good news! Now that they know what to do for me!” , it is just not the case. Rarely is the news all that good or all that simple.
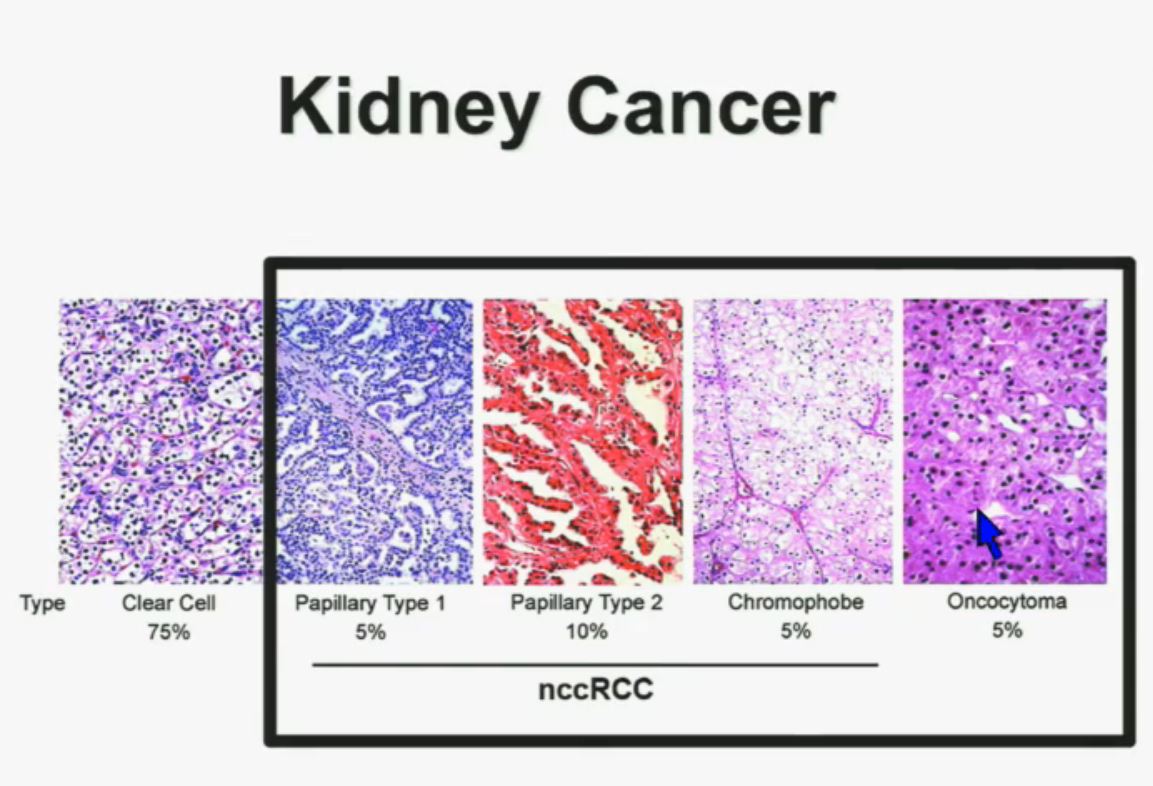
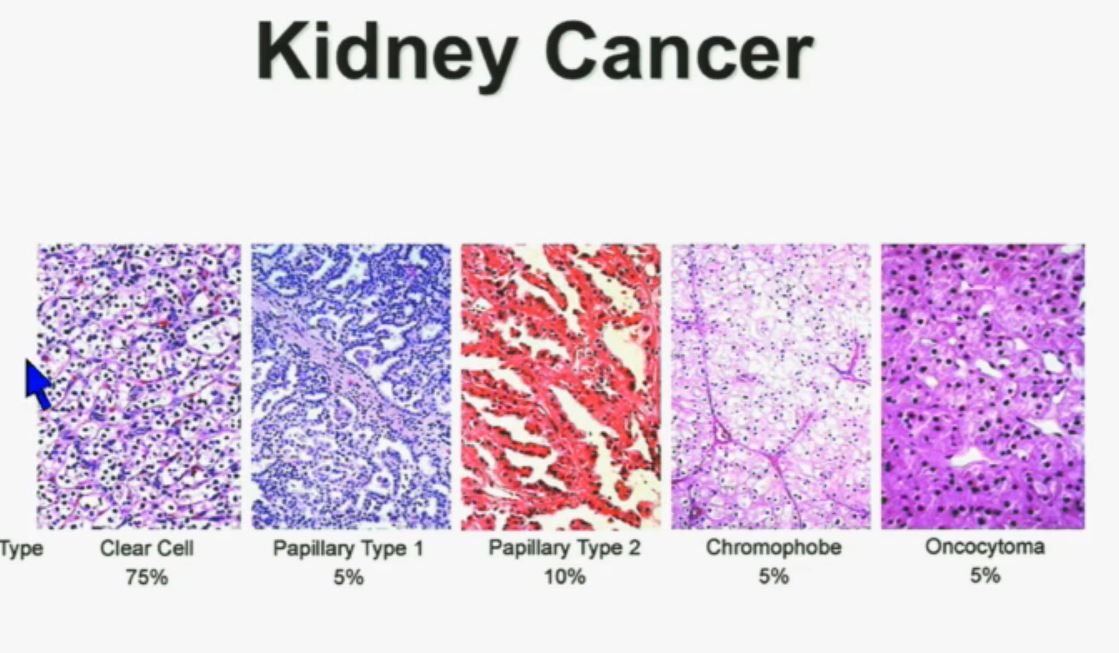
Let’s back up here and lay the groundwork. Clear cellRCC, or ccRCC is the most common of about 10 RCCs. They all land in the kidney, but can vary widely. ccRCC may be about 65% to 85% of the cases of kidney cancer, with the rarer non-ccRCCs making up the rest. Maybe 15-35% of the RCCs are considered rare, with the most common Papillary Type I, Papillary Type II, chromophobe, clear cell papillary, collecting duct/Bellini’s, medullary, translocational (not to be confused with transitional, etc, etc.) and to make it still more confusing, unclassified RCC. But when the most common is described as either 65% of the whole or 85% percent, you have to question if there is clarity in that category!
Clinical trials for RCC have usually only included patients who had clear cell. The reasons are simple; it is the biggest group, the patients can be more readily found, and that is the largest group in need of the medications. But the patients with nccRCC are really also terribly underserved. Back in the day, none of us had many options beyond surgery, so little distinction was made. The prognosis was grim all around, once the cancer had spread.
But the new world of precision medicine, in its name alone, reminds us that the meds need to be developed more precisely, that they be given to the right patients at the right time. The general crap shoot or “wild-ass guessing”, as a friend says, still remains. The latest (not necessarily greatest) group of meds are the newish immune therapies. You have seen their ads, no doubt.
One of those is Opdivo or nivolumab, its research name. It tries to unblock some of the inhibiting mechanisms that prevent the immune system from doing its job, but it has been tested in trials only with clear cell patients. BUT, that does not mean that only clear cell patients are being prescribed the meds–this, thanks to the slightly wild west of the US medical system, that can truly go beyond the FDA approved medication guidelines.
This study, which will be formally presented at ASCO in June, 2017 was announced with the headline above, “Nivolumab Shows a Substantial Objective Response Rate in Refractory Non-Clear-Cell Renal Cell Carcinoma”. The researchers are NOT in charge of the headlines, so we must dig deeper and see what this study really means to the patients with nccRCC
I tried to sort out what it means–or does not mean. My quick review is that it does not give a great deal of clarity to the majority of those nccRCC patients. A more complete report may improve upon this. Based on this link, I offer the following:
http://www.practiceupdate.com/news/16132/67/10?elsca1=
“I am always concerned that these new study reports are characterized carefully. They are always more complex and incomplete than I would like. A patient in a forum says this tells of ‘good’ responses, and especially so for the non clear cell group, but s does ‘good’ really mean generally a benefit to those rarer nccRCCs? Until a fuller report emerges, I can only note the following:
There were 23 patients, from three centers, with a median age of 59. Surprisingly 30% were African -American. This may tell us that there are more African-Americans with the rarer non-clear cell RCCs, or could reflect the local population of the three centers. Only 23 patients and with a mix of diseases will never meet the statistically critical requirements to reach the level of excellent evidence–but it may be all we have at this point.
All 23 had non-clear cell, but nearly half had ‘unclassified’ RCC, quite a high rate. Usually that is considered to represent between 1 to 4% of renal tumors. Most of the rest were papillary, but they generally make up the largest percentage of nccRCCs. No distinction is made here between Papillary Type I or Type II, which are really quite different diseases. Papillary Type I and II are the most common of the uncommon, non clear cell RCCs, and are readily distinguished from each other. This would be valuable info, and wonder if this was noted in the fuller report.
Only 3 of 4 patients had nephrectomies before the trial treatment. Were 1 of 4 patients too sick to be given the standard of care of surgery or were their doctors unaware of that? How does this affect the study, and were the no nephrectomy patients from one center or with one subtype? We do not know the reason for this high rate of no surgery, and at a time in which it is clear that the removal of the tumor is a great benefit to the patient, metastatic or not.
Two-thirds had metastatic disease at the time of diagnosis. Of the total 23, 74% had a prior treatment, mostly Sutent or Votrient. Of these patients with prior treatments, 26% had TWO such treatments. Thus these patients had already received treatments that were not directly approved for their subtypes. This is not too rare in the US, where we have greater leeway from our prescribing doctor than do patients elsewhere. But how does this fit in with the relatively low rate of nephrectomies?
This report does not say how quickly they were treated, i.e., how long from initial diagnosis until treatment with Nivolumab?A patient with Papillary Type II found to have no metastatic disease at the time of diagnosis, but who received a nephrectomy, was monitored for a year or so, then went on one or more systemic therapy is quite different from the patient with an unclassified RCC, metastatic at the time of diagnosis, not given a nephrectomy, though treated quickly with Nivolumab. What can be learned when there are such wide variations in just 23 patients that would be helpful to the Papillary Type 1 patient?
The follow up period was a median of 6.5 months, which seems very short, especially when the median Progression Free Survival of the responders was 4.2 months. The median OS is not given. That certainly may reflect an ongoing study situation, or a failure to provide a longer period of follow up.
As to objective response, 6 of the 21 evaluable patients (29%) had a Partial Response, which would likely be a 30% reduction in metastases. Another 4(19%) had Stable Disease. Two of the 23 patients died, but not from the treatment. (Assume that had to be due to the disease, but certainly indicates that for nearly 10% of the patients, this was not at all effective.)
When the final analysis was done, nine patients were still receiving Nivo. Newly recruited patients might still be in treatment at that time, but those recruited earlier may have gone out of the trial at the same time. It is important to not that Nivolumab treatments were stopped in three patients due to intolerance, and six more had postponed treatment, i.e., 9 of the 23.
Certainly we need to find meds which create responses for nccRCC patients. However, I am concerned we draw any certain conclusions from this study. Indeed, it is “good” to know that the treatment was tolerable for the majority of the participants, but not so good to read that 6 of the 21 patients had to postpone treatment, and three were removed from treatment due to intolerable/toxic side effects. We also do not know which subtypes seem to have shown responses, which would have been qutie easy to report. Did the group with Papillary Type II do generally better that the majority “unclassified” group? No answer from this stury report. And in the back of my head, I keep wondering why in the world there were so many unclassified patients in this small study? Was there a standard pathology review, or could these patients been misdiagnosed by one pathologist. Typically there is a single pathologist which can standardize the reporting. Were all these patients properly diagnosed?
Just wishing there were greater clarity and hoping to get a fuller report, post ASCO.
Without a doubt, the ‘good’ that comes from this sort of report begins with the recognition that the nccRCC group is underserved by the research community> They probably have the poorest outcomes, rarely have a clear diagnosis, and must wait for the ever popular “further research is warranted.” But all must be aware that these very small observational studies must be reviewed very carefully for what they show or do not show. Again, one to watch at ASCO, but not enough to make a major change in treatment for any one with a non clear cell RCC.
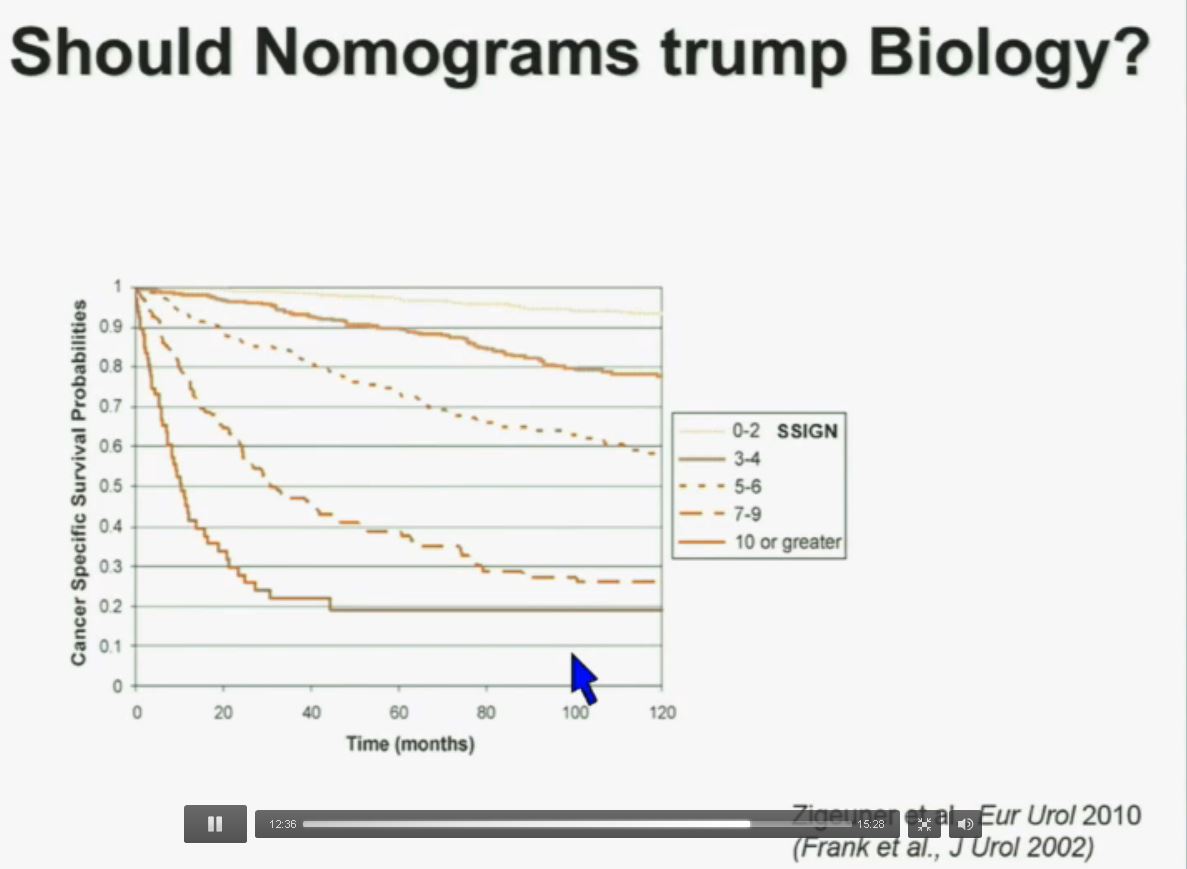
PS. Does your doctor know that there are at least four subtypes of clear cell–the big ‘common’ group–which have clearly different survival patterns? Thought so.



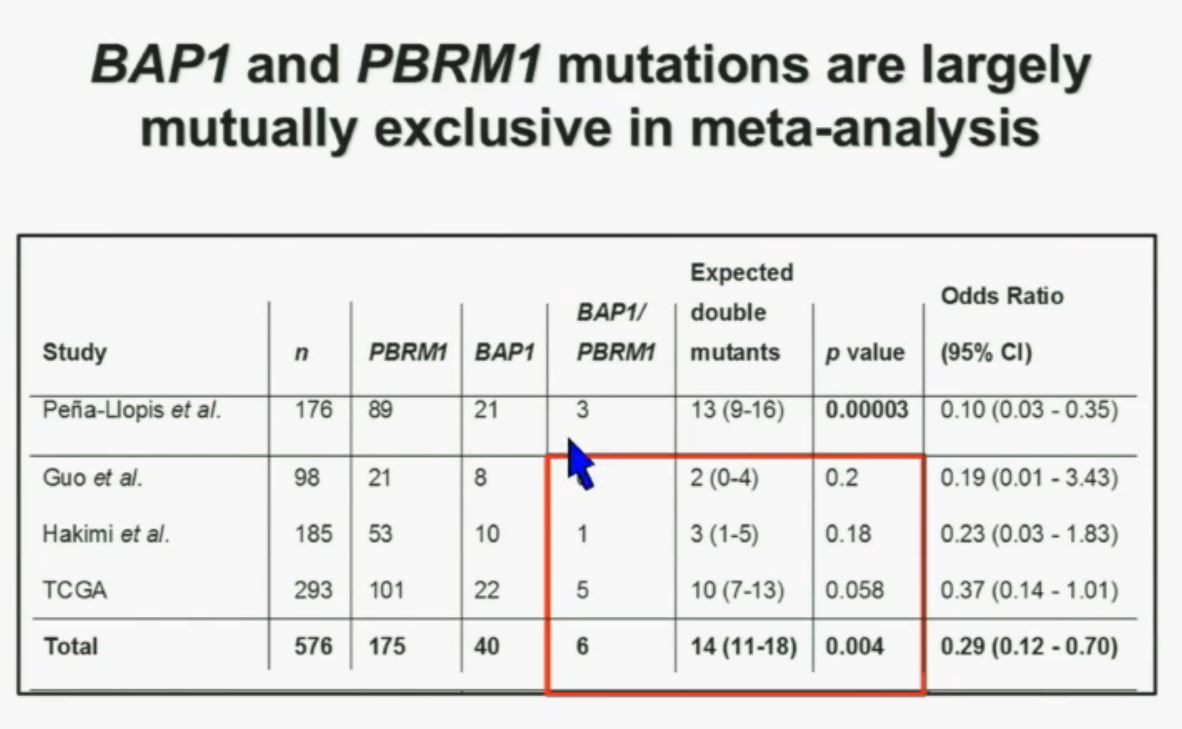

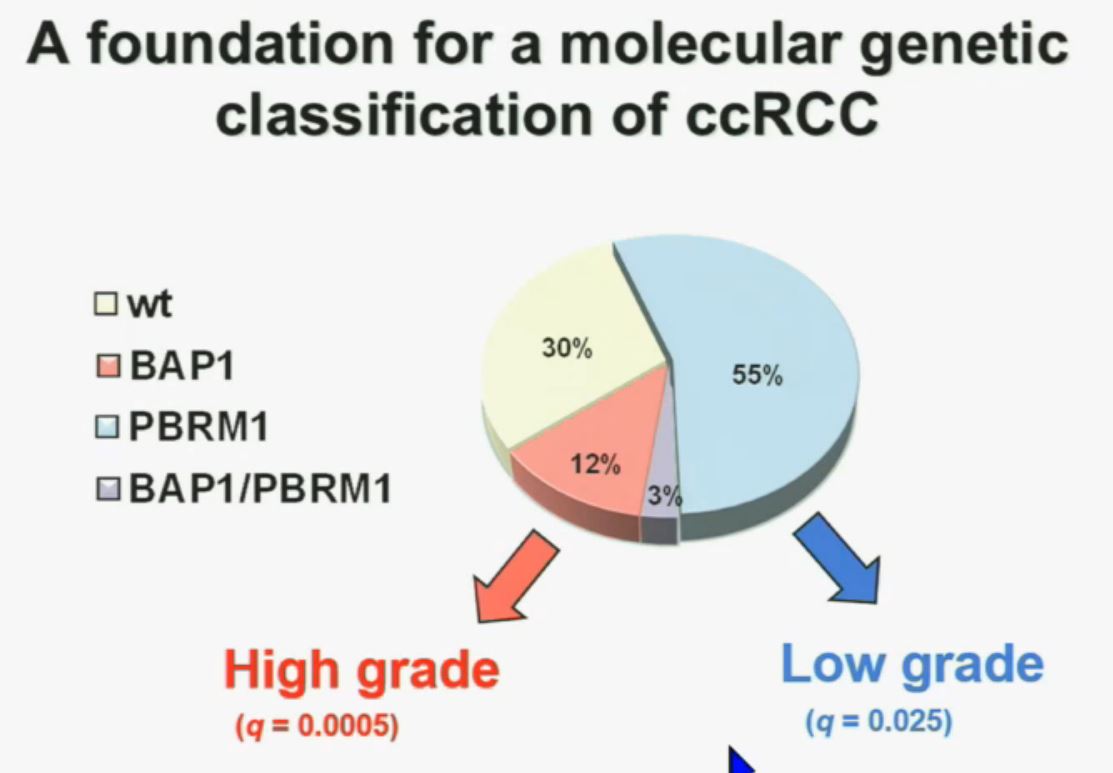
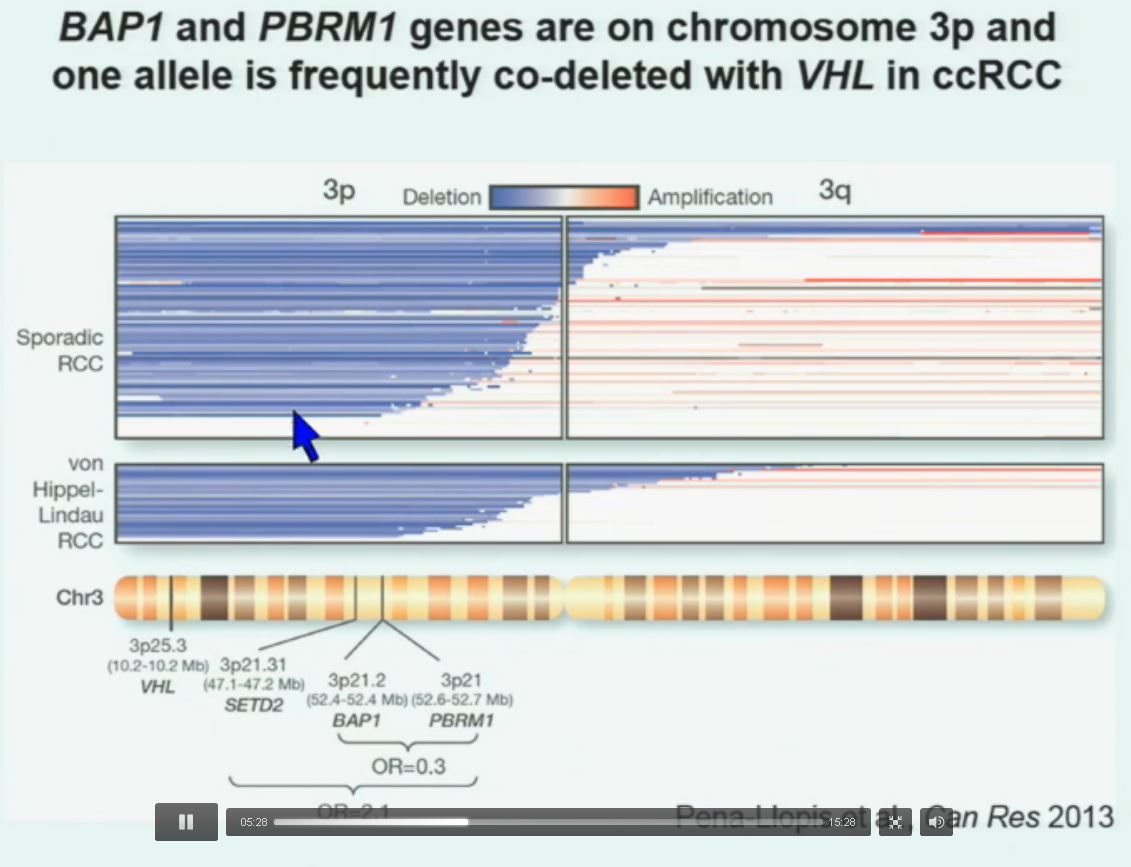
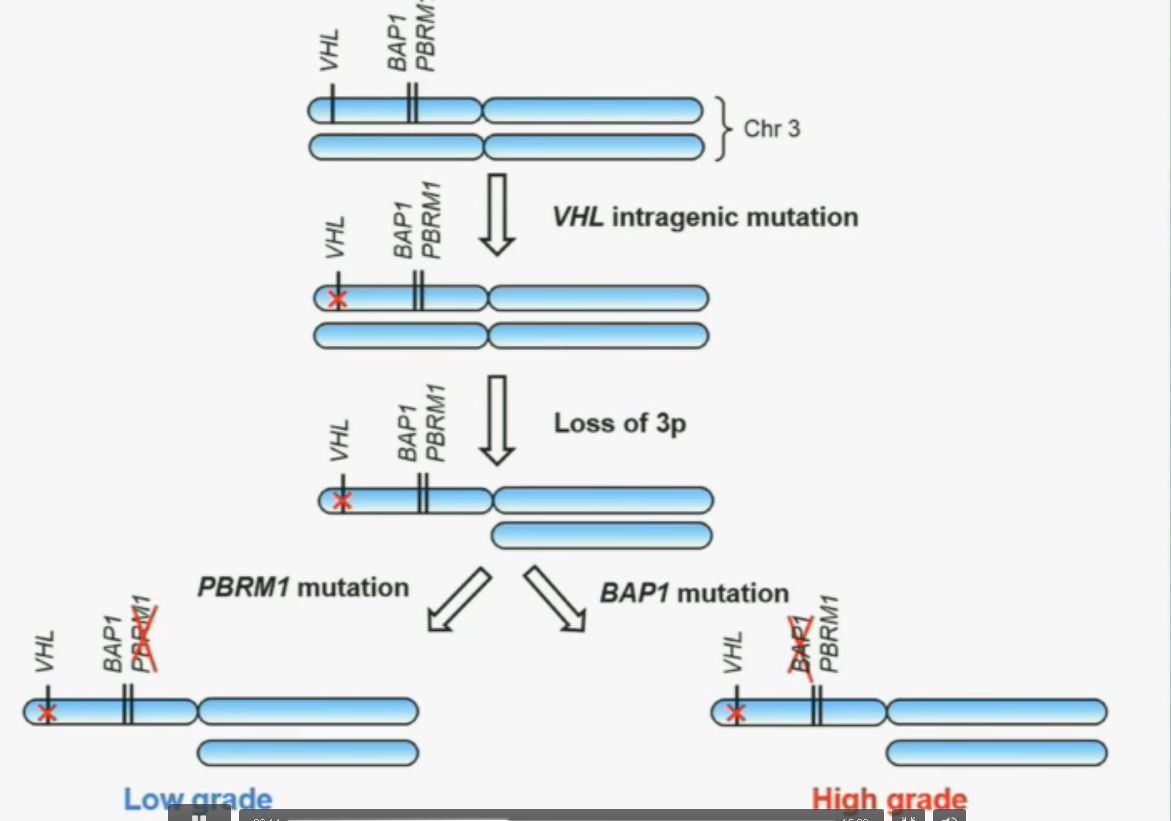
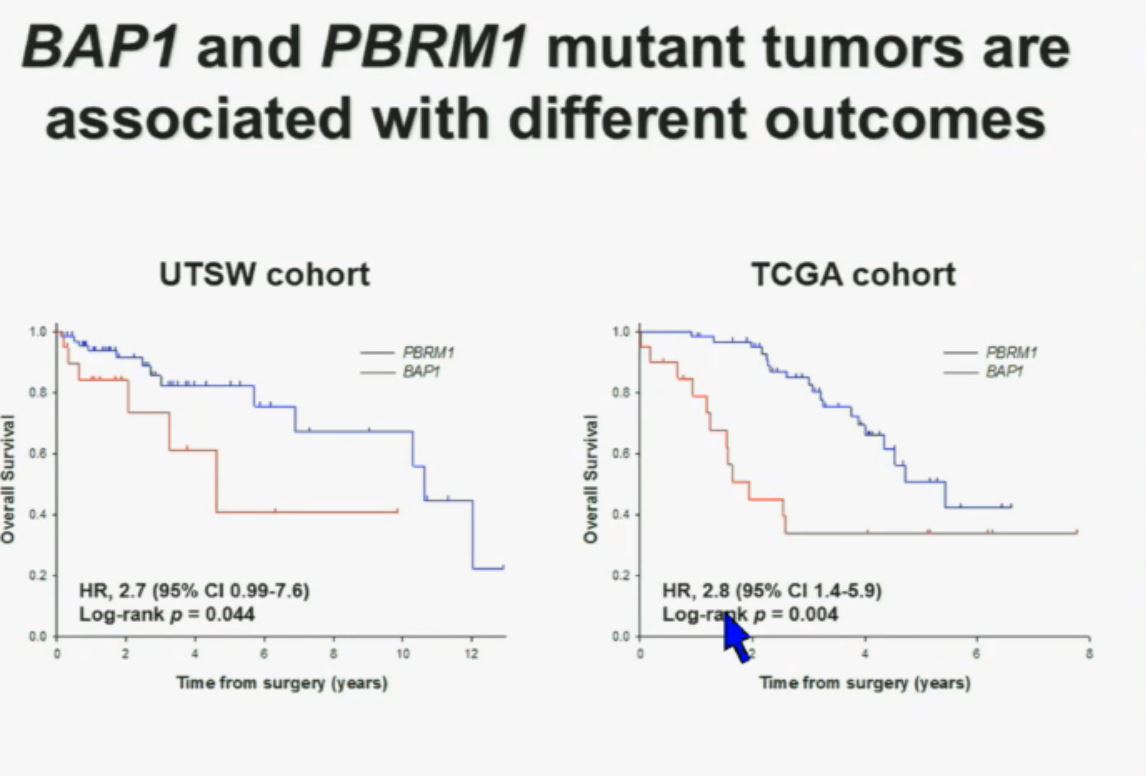

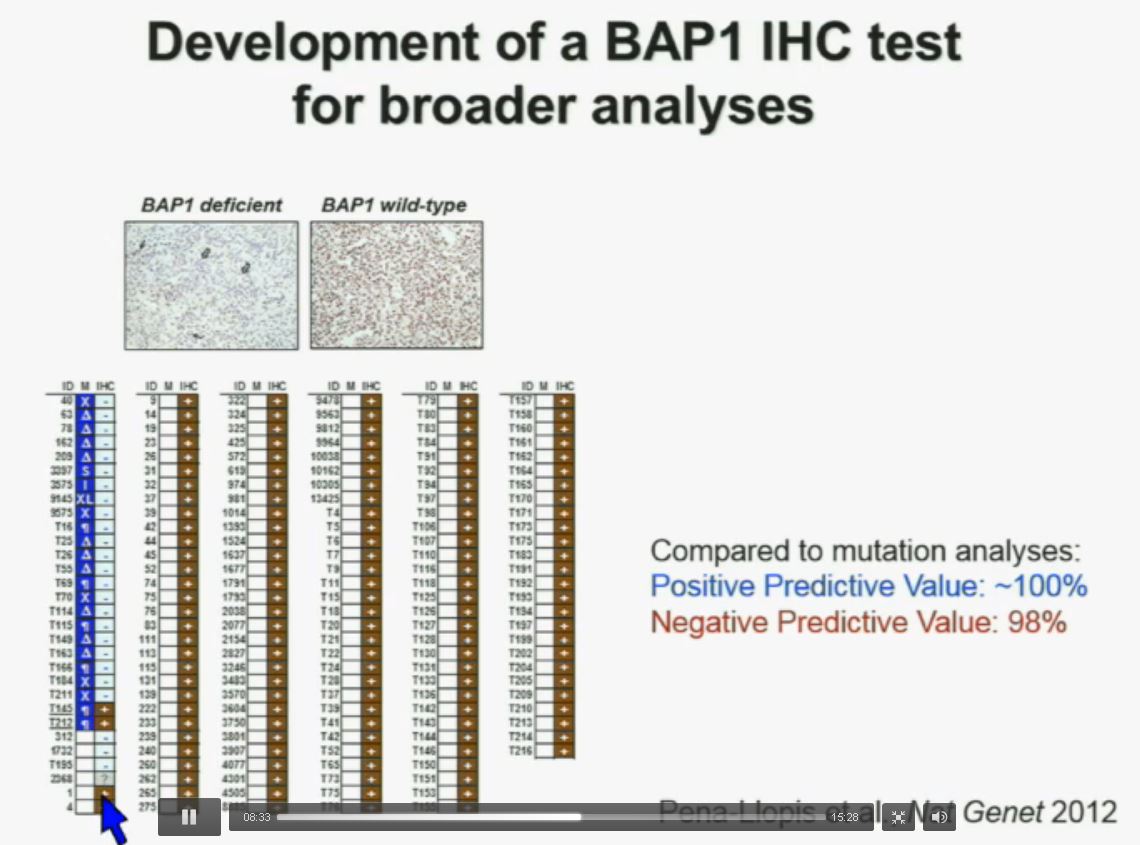
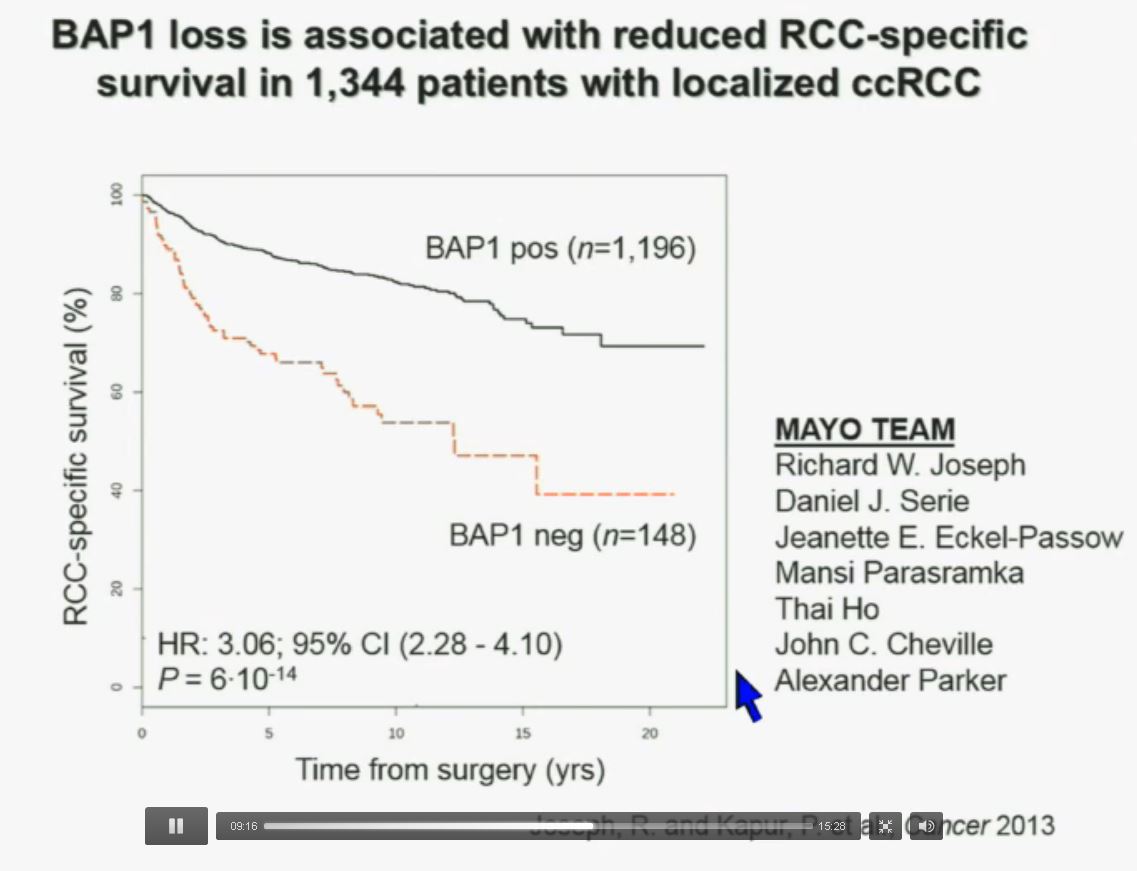
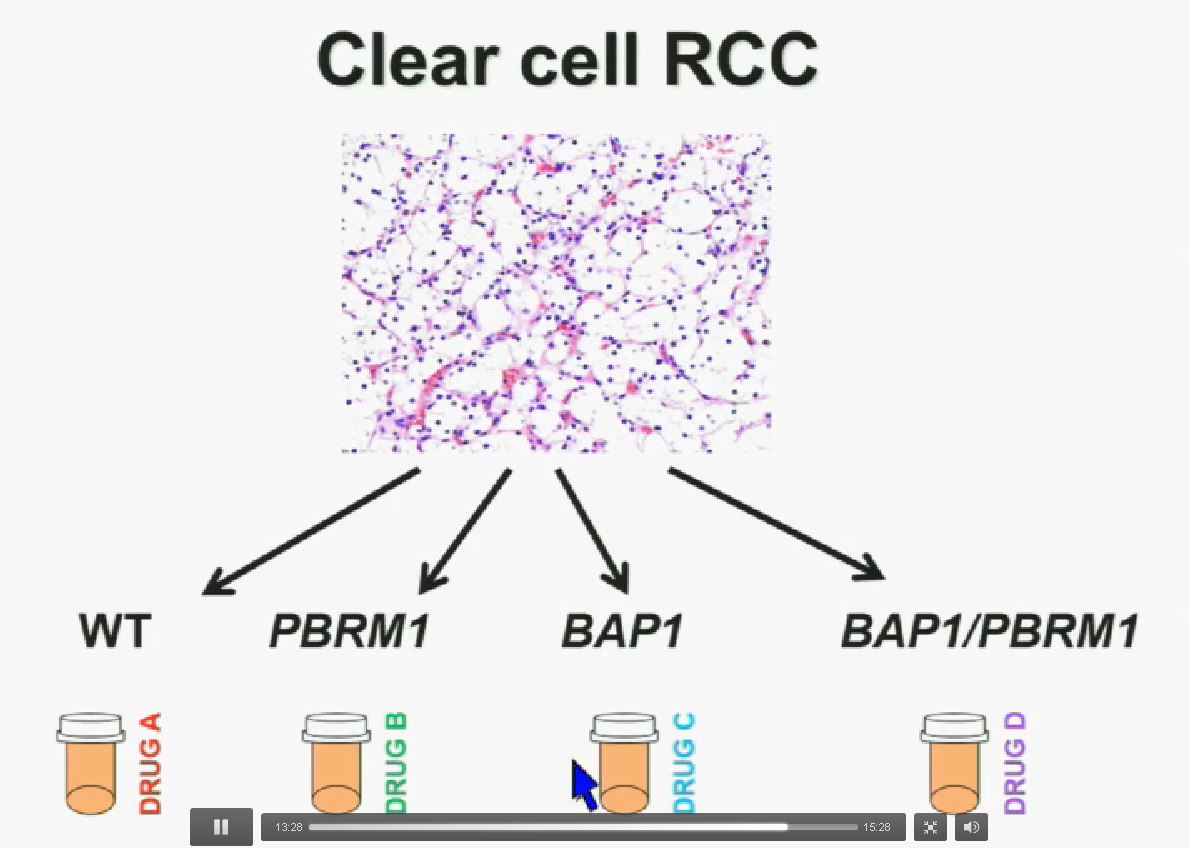
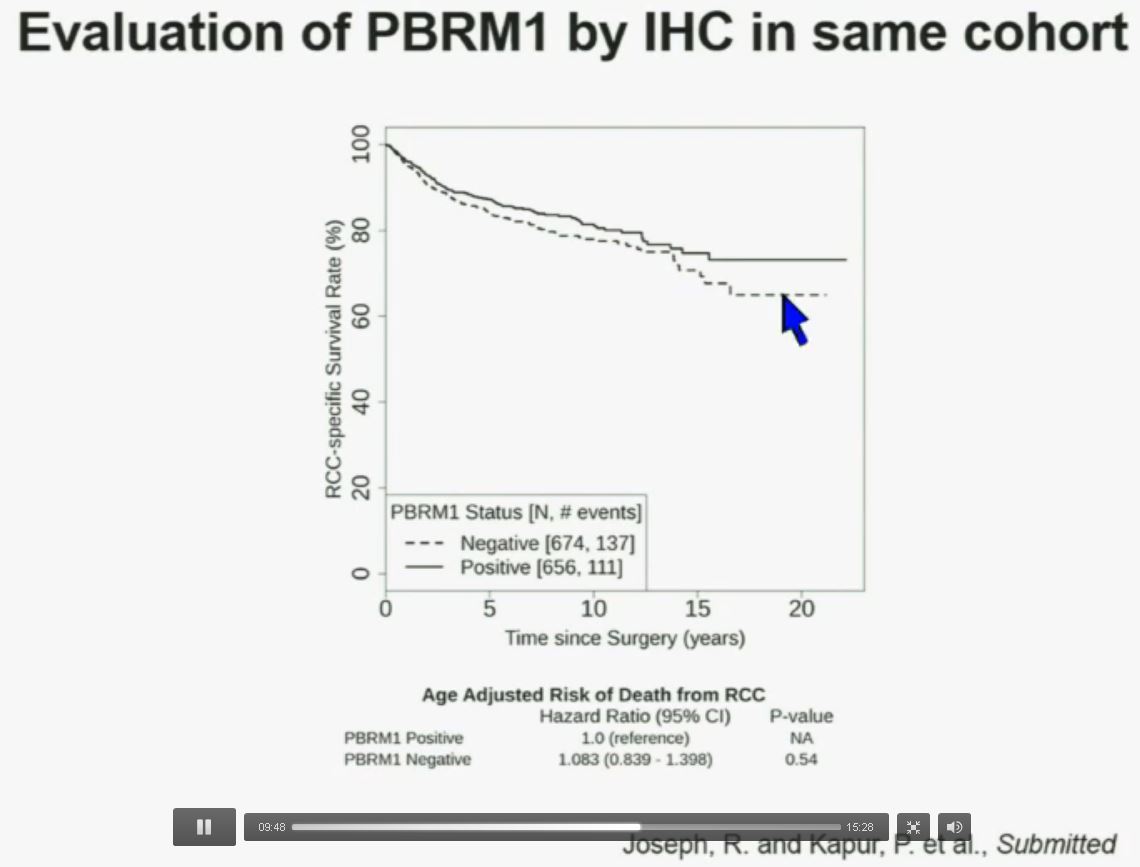 Now in the same cohort we looked at BPRM1, which like BAP1 in a two-hit tumor suppressor gene, and we find no significant differences.
Now in the same cohort we looked at BPRM1, which like BAP1 in a two-hit tumor suppressor gene, and we find no significant differences.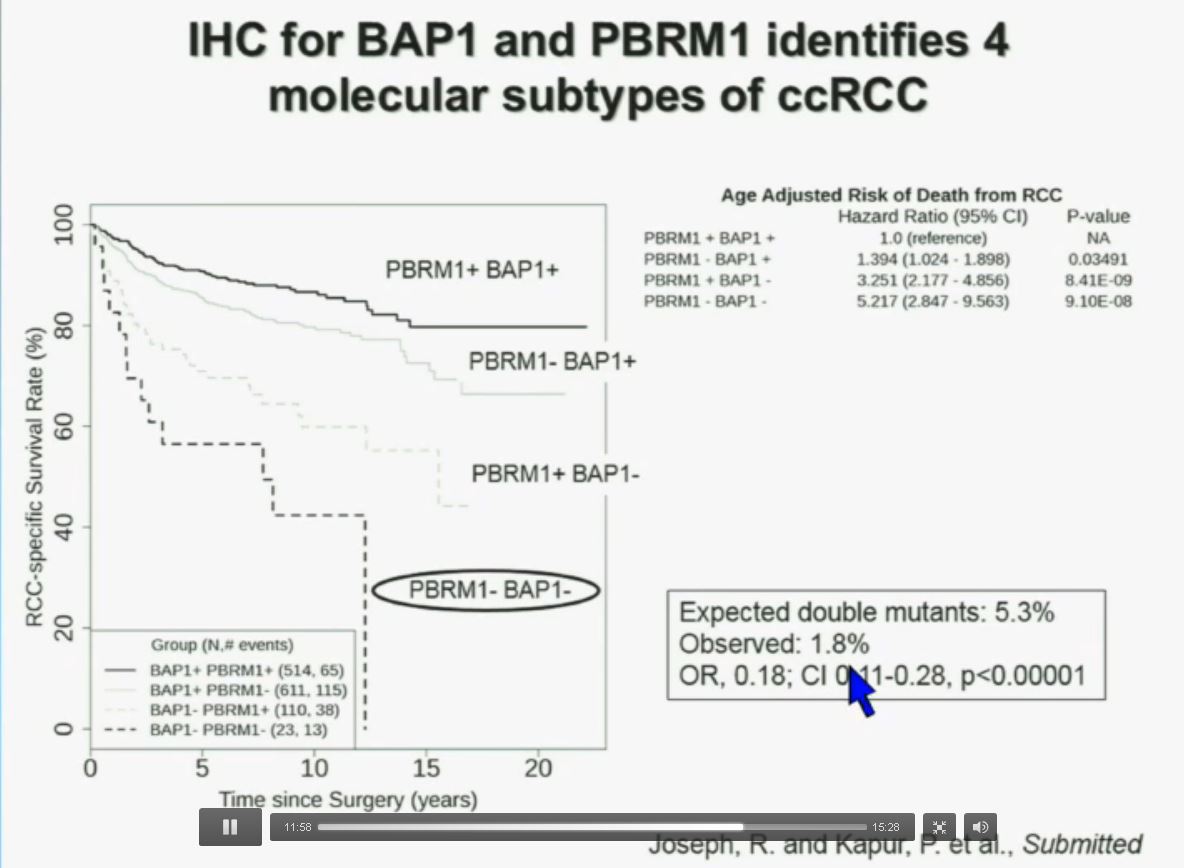
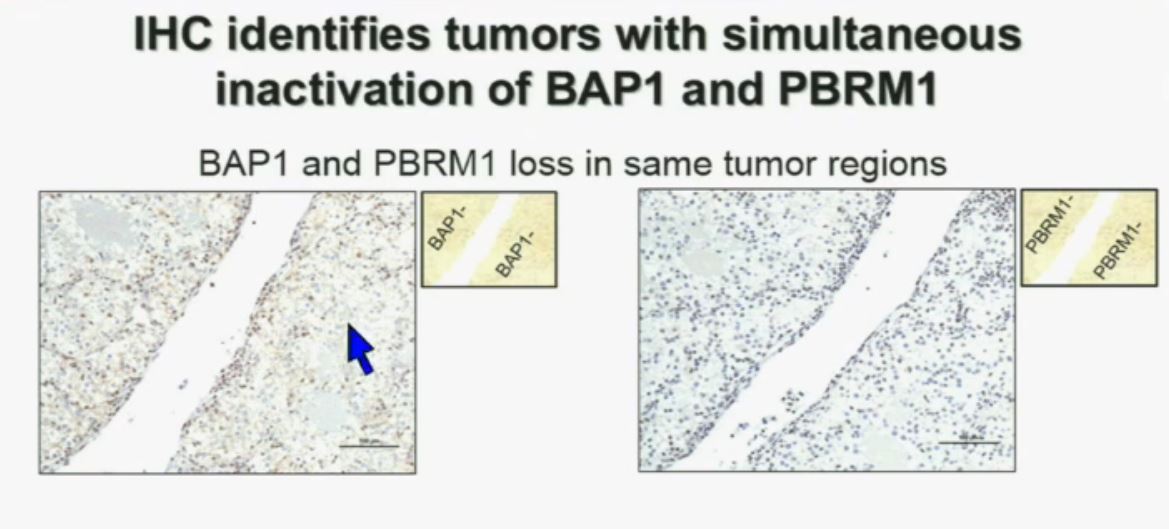
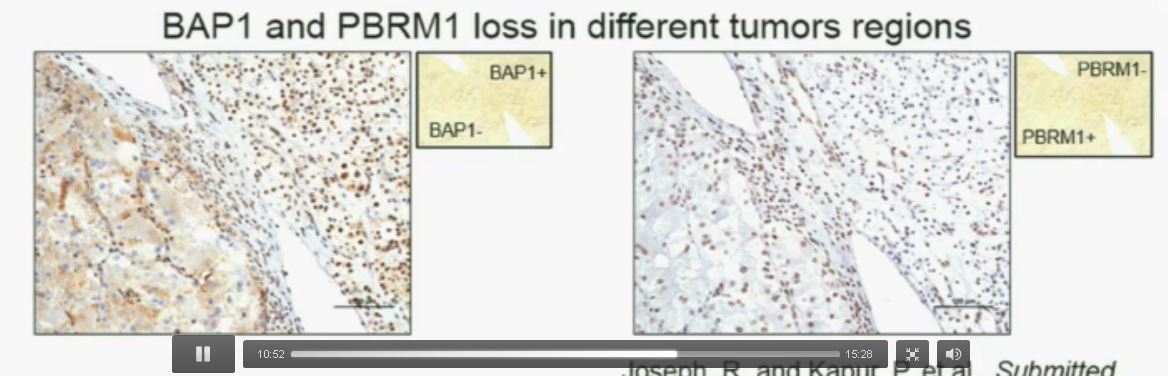 Slide C
Slide C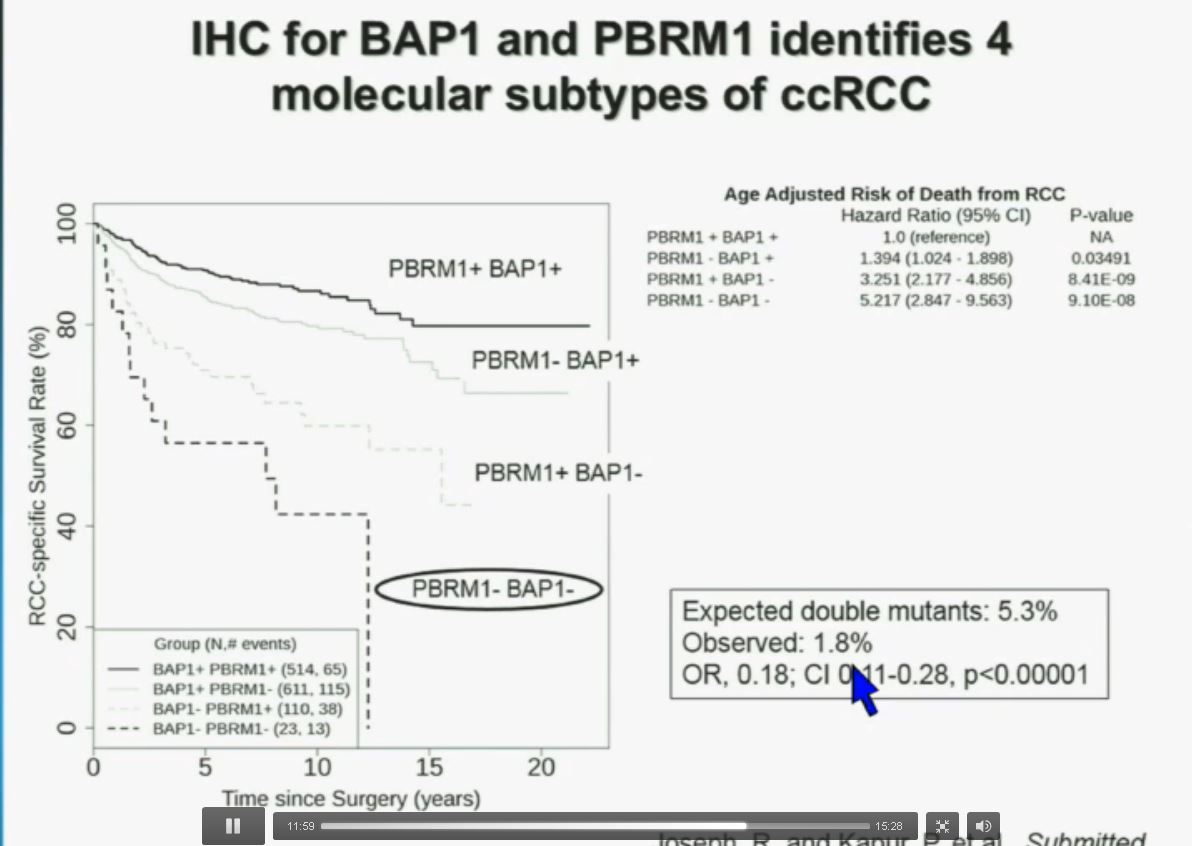 Slide should include quote “BAP1 and PBRM1 do NOT predict outcomes independently of SSIGN”
Slide should include quote “BAP1 and PBRM1 do NOT predict outcomes independently of SSIGN”