For years, doctors have debated one another about what to do with little kidney masses, i.e., “small renal masses or SRMs” in doctor-talk. Patients hear this debate only when it applies to them, or if there is some hysterical headline about tumor cells escaping the tumor because a biopsy was taken. What is the reality, and what value does this have to the patient?
First of all, most large tumors/masses on the kidney are found by CT or other imaging. In the kidney tumor world, large can mean
For years, doctors have debated one another about what to do with little kidney masses, i.e., “small renal masses or SRMs” in doctor-talk. Patients hear this debate only when it applies to them, or if there is some hysterical headline about tumor cells escaping the tumor because a biopsy was taken. What is the reality, and what value does this have to the patient?
First of all, most large tumors/masses on the kidney are found by CT or other imaging. In the kidney tumor world, large can mean anything over 3 to 4 centimeters in size. Taking out my inch ruler with its handy centimeter imprint, I see that is just over an inch to about 1 1/2″ in size. Might be the size of a walnut to so–still doesn’t belong there, nor sound too insignificant to me!
Nevertheless, that is a Small Renal Mass, and is not even considered for treatment by some doctors. Our newer and more frequent imaging can find tumors of this size, long before they would be felt by a patient. They may or may not ever grow much larger, or do so very slowly. In fact, about 25% of these SRMs are not cancerous. Rather reassuring, except for the obvious conclusion that 75% of them are indeed cancerous! Size doesn’t matter in this case! Add to that the possibility of that benign mass may continue to grow and mutate/change over time. Its benign character may not remain benign.
Some 10% of these masses may subtypes of RCC which rarely grow,and imaging cannot determine that. The patient may be too old or ill for surgery at the time of discovery. A rush to surgery may not be appropriate but can a biopsy answer some questions–and is that dangerous?
Some have raised a concern that inserting a needle into the mass to get cells to examine is inherently dangerous, and could release cancer cells into the body, especially along the track of the needle. Of course, any mass large enough to be seen is likely already sending out cells in the course of its growth. The chances of any such cell becoming an established tumor is incredibly small, but every metastasis got started from some ambitious and lucky cell landing on a fertile spot in the body.
The reality is that there have been very few cases of obvious tumor seeding along the needle path, as a biopsy is taken. And these biopsies can be very helpful in determining whether or not the mass is cancerous. Thus a biopsy should not be avoided, if there is a question as to the nature of the biopsy or if surgery is considered inherently dangerous.
But does the biopsy give all the answers? Unfortunately, it does not, and especially since the typical biopsy will not differentiate between certain of the newly-discovered subtypes of clear cell renal cell. In short, some clear cell tumors may be destined to be more aggressive, while others may be very slow-growing. This can be analyzed only by a molecular review of the cells, which is not done typically. Thus, even a biopsy–now thought to be far safer than in earlier years–may not provide a solid guideline for the next treatment. Getting the molecular analysis, as described in a previous blog about ccA and ccB variants of clear cell RCC, will become essential for patients in the near future. Or so I fervently hope.
“Now what?” may be the first coherent question a newly diagnosed cancer patient asks. Maybe the smarter version of that is “What–when and why?” And your doctor had better have a good answer, as to the treatment, the when and the why.
We cancer patients usually get surgery “first”, even when the disease has spread. Primary surgical strike and then a clean-up operation, in the ‘war on cancer’ parlance, we think–when we can think. “But which is the best and first clean-up approach?” we must ask. “What works the best? What can I take with my other health problems? Where does surgery or radiation fit in this scheme? What does the doctor favor and why? Where do I get this treatment? And then what?”
Treatments and their sequence are often chosen with little reliance or clarity as to the data. But some light was shed today at ASCO (American Society of Clinical Oncology). It released a comparison of the sequencing of High Dose Interleukin2 (HD IL2) and of targeted therapies for metastatic RCC. Which should come first?
It shouldn’t be a high-stakes gamble to choose a medication, as no one can guarantee any results–with any of the meds. You take a chance with any drug, so which do you start wi We may be closer to a logical approach in sequencing these drugs. Sequencing of these highly different medications has measurable effect on overall survival (OS)—and to patients’ lives. That sequencing is critical and certainly can extend life, even when treatments fail, as they so often do.
A retrospective study of 97 US patients who received HD IL2, before or after a targeted therapy was just presented at ASCO. These patients were followed for a median duration 37 months–half more than 37 months, half fewer than 37 months. Of that group, 22% had either a partial (14%) or complete (8%) response to HD IL2. (No specifics as to what was a “partial” response, perhaps a 30% shrinkage of the total tumor burden). In addition, another 24% of patients had Stable Disease(SD). Thus, nearly half of these patients benefited from having had HD IL2.
Stable disease is better than progressive disease, as any patient knows, though it was rarely measured in older trials. Though we patients really want a cure, we do want to be around for the next treatment, to have a surgery or ablation to remove the “stable” tumor, or to try another medication.
Of these 97 patients, 82 received HD IL2 before any targeted therapy. Another 15 patients had HD IL2 following a TKI therapy. That timing made an important difference. HD IL2 followed by the TKI, showed a median Overall Survival (OS) of 61.8 months. The OS of those with the TKI before the HD IL2 was 48 months. A median, not an average, so half lived longer, half lived shorter than the quoted medians.
A pre-2006 NCI (National Cancer Institute) series showed a 19 month median survival for HD IL2 alone, and a similar 19 months for the use of targeted therapy alone. Using the two in sequence dramatically improved OS, especially when HD IL2 was first line of treatment. Obviously things have improved, though it can be very difficult to compare older trial data, as so many variables are different–including the type of RCC the patients had as they entered the trials.
Several points can be made from this study. First, no therapy should be examined only as to Complete or Partial Response. Stable Disease also adds to Overall Stability. To stop the tumor from growing, even if for a period of time, is valuable to patients and can prep them for the next anticipated treatment. Sure beats tumor growth!
Second, therapies should be chosen to maximize their impact on the overall survival of the patient. Some patients will naturally be precluded (or delayed) from surgery, or taking one drug due to existing co-morbidities, due to heart disease or liver damage. For those post-op patients, likely to tolerate the side effects of HD IL2, it should be given in a first-line setting.
The most critical variables that impact patients are the recommendations and expectations of the physician. Most patients are not even told about HD IL2 treatment, or it is dismissed casually as “not for you”. Others are told to wait until more mets emerge, with some weird theory that waiting for more trouble is a good thing! Many nephrectomy patients are not monitored post-operatively, despite the risk of mets. This is surely an indicator of the lack of knowledge by urologists. Still others are told that the disease has spread, and that nothing can be done–also untrue.
The rarity of RCC and its variants leaves most physicians unaware of all options in the field, and how to any one might suit for a particular patient. Most oncologists to whom patients are referred, have little or no experience treatmenting for RCC, or may not access to academic centers for support until it is too late. Even the pathology of the primary tumor and later metastases may be questionable, adding to the challenge of care.
With the dramatic changes in the RCC field, this is to be expected—but not tolerated. The patient may have to provide his physician with the data that can extend or save his life, which is a sad but realistic commentary on the field today.
Most people are not surprised that there is no ONE thing called cancer. Tumors in all the organs or invasive cells in the blood or bones are referred to as cancer, but start when cells go wrong, whatever the cause. As soon as you are told you cancer, whatever it, the quest begins to find out exactly which cancer it is. With kidney cancer, or its more melodious name, renal cell carcinoma, there seem to be endless variations on what may be called kidney or renal cancer. To treat it requires a very careful analysis of what is really is, starting with the pathology of the tumor when it is biopsied. With kidney cancer that biopsy is usually done after surgery for the tumor. That biopsy will describe the shapes and type of cell in the tumor, which can be mix of types. And then the real work begins.
A recent article in “European Urology” reviewed the mix of HEREDITARY renal cancers, those that arise due to one’s background. More common are the “sporadic” kidney cancer that could arise out of the blue or in response to some environmental toxin. There are ten Heredity Renal Cancers, or HRCs. My goal is to alert the reader to the possibility that his cancer might be one of these. This would affect treatment, and may suggest the need to test family members.
If you have kidney cancer or RCC, you may be familiar with “clear cell” or “papillary” to refine the description of the cells in the tumors. This may not be the whole story, as those HRCs—the hereditary kinds—may manifest a mix of ways, including as clear cell or papillary histology.
The most common HRC is Von Hippel-Lindau (VHL) disease, with both benign or malignant tumors. RCC can be found in a 24-34% of VHL patients, appearing at mean age 39 years (far younger than non-heredity RCC), and often with multiple tumors and in both kidneys. Cysts which appear not to be malignant must be watched–they have the potential to become malignant over time. Generally they are managed based on the size of the largest of these lesions. Clear cell RCC is the one VHL-related subtype.
Hereditary papillary renal carcinoma (HPRC) is rarer, and typically occurs later in life. Papillary tumors are the only phenotype with HPRC, and patients often develop numerous tiny tumors, 1000 or more. These tumors are considered type 1 papillary renal cell carcinoma (pRCC) with a low nuclear grade, monitored with CT scans, and some do metastasize, though this is rare. The MET gene is implicated in the growth of these tumors.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is newly identified as a HRC. Rarely do patients develop RCC, but are susceptible to developing multiple leiomylomas, which are generally benign. When there is early onset of HLRCC, then RCC is found in about 20% of those patients. These tumors can be aggressive, and about 2/3 display a papillary pattern. Such tumors tend to be hyper-vascular.
Birt-Hogg-Dube (BHD) syndrome is quite rare, about 1 in 200,000 people, and thereby likely under diagnosed. This raises the risk of developing kidney tumors, which occurs in 25-35% of BHD patients, and at mean age of 50. These tumors have varying histologic subtypes, generally chromophobe RCC or hybrid variants. And there can be variants in the same tumor or within the kidney. There is a risk of metastases, though rare. The characteristic skin lesions of BHD syndrome are not malignant.
Even more rare is Tuberous Sclerosis Complex (TSC), which can manifest itself in renal lesions, cysts and occasionally, RCC, the latter at a young, average age 28. Neurologic complications can accompany this syndrome.
SDHB-associated paraganglioma/phaeochromoytoma is another heredity condition which may give rise to a mix of renal tumor, including clear cell RCC, chromophobe RCC and oncocytomas, i.e., a mix of histologically different types.
An HRCmay be suspected in patients with a family or individual history of renal tumors, in the instance of both kidney having tumors, or one kidney having multiple tumors or in early-onset renal tumor, i.e., under 50 years of age.
Clinical diagnosis can be further refined by genetic testing, and thorough review by an experienced uropathologist is fundamental to the diagnosis. First consideration would be a VHL analysis and genetic analysis of SDHB and FLCN genes, as warranted. Patients with type 1 papillaryRCC should be considered for MET analysis. The presence of clinical symptoms related to any of the syndromes will guide the gene screening. Testing on family members may well be warranted.
With these cancers, it is possible to have multiple lesions and affect both kidneys. Thus, treatment should preserve renal function and control the risk for metastases. Use of ablation to retain maximum renal function may be preferable to partial nephrectomies, for example.
Though these heredity renal cancers arise in a different manner than the more common sporadic RCC, the study of the molecular pathways provide some insight into new therapies for those patients as well. Thanks always to those researchers who help in this struggle for information, as that is essential to provide treatments.
One of the many challenges in fighting kidney cancer is knowing where it all got started. This research indicates that a gene gone astray, the MET gene, is part of the problem from some patients. It is this type of study that will change the treatment for us, as there will be greater clarity as to the “target” to be reached by “agents of change”. (Nothing political intended, but seems to work here!) Especially of interest to patients who have the variant of papillary RCC, about 10% of us.
MET Variant as a Prognostic Marker in Clear Cell Renal Cell Carcinoma
Dr. Ari Hakimi of Memorial Sloan-Kettering Cancer Ctr.,New York USA
ASCO GU Congress 2014
eCancer reporter Peter Goodwin’s questions are in italics. Where I was not certain of the lecture, I added a (?) to show that. Link below to the actual lecture.
Ari, you have been looking at prognosis or prognostic features, or actually, molecular features of renal cell carcinoma. Can you tell me what you were doing in the study you’ve just been talking about?
There was a great paper that was published by the people at Harvard and Lancet (?) Oncology last year. It found for the first time a prognostic marker that was associated with poor survival in kidney cancer, a molecular marker. This was a variant, a normal variant in the gene, the MET gene. In that study they had several hypotheses they generated from that study, but they didn’t really have enough genetic data to try to figure out what was going on here with this variant in the genes. So what we did was, we took that same concept, that same variant, in the Cancer Genome Atlas Study, which has both patient information and then a host of genomic information. We tried to validate their finding and explore the biology of that marker.
It sounds like a needle in the haystack but you’ve but you become quite familiar with this variant called RS11762213. What you know about it so far?
We know about that it is a variant in the MET oncogene, a very important gene in a lots of different cancers, particularly in papillary renal cell cancer. It’s a gene not thought to be very important in clear cell renal cell carcinoma, but we found that it is, and we explored the variant in an exonic region of the gene–meaning the coding region of the gene. Because the variant is in a coding region of the gene, we thought it might be more than just a prognostic marker. It might also have some biological implications.
Biological implications? What sort of biological implications?
We think the marker may be; we figured out through computational methods, exploring the TCGene data(?) that it might be in the region of enhancement, meaning the variant leads to higher activation of the MET oncogene. In turn, this might explain why these patients have a poorer outcome. It might also have potential therapeutic implications.
So what have you found so far then, about the level of additional risk if you happen to have this variant gene?
Great question. We took about 270 patients from the cohort who had available information. We genotyped them, meaning we determined what percentage of these patients had the risk variant, which is about 10 %, consistent with prior studies. We showed that when these patients had that risk, in addition to the current prognostic features, they had about a 3-4 fold increased risk of cancer-specific death, or tumor recurrence after surgery.
That’s really quite powerful! Am I right that there wasn’t any clear kind of molecular feature to give you some help in the past?
Until this study, which was published last year, there were really only tumor features and patient features that were associated with poor survival in kidney cancer. This is the first study that really showed, that they published last year, to show in two different cohorts that had a molecular feature that added to the prognostic models. We showed, augmented their findings, that if you took the best current prognostic models and stratified patients, added to that model, meaning it improved the predictive accuracy of even the best post-surgical models that are out there.
You are looking actually disease mechanisms–mechanisms of cancer production. You established prognosis, but what about predicting response to therapy?
That’s a great question. Our goal now– that we’ve established that this is a valid biomarker, truly multiple cohorts now showing this marker can stratify patients for aggressive behavior, we can now explore—hopefully–whether this has therapeutic implications because it is in a gene that is a known cancer gene. Because there are multiple drugs that target this gene, and because we think that this variant that is activating this gene, it stands to reason that an inhibitor for these patients with this variant might work. These patients might have another option.
So theoretically a new drug which is an inhibitor for this variant might work. What about existing cancer drugs? Do you have any ideas about if any these do influence that variant?
We don’t know yet. We are trying to find it in cell lines, meaning cell lines that are derived from tumors that are used in the lab, to see MET inhibitors that currently exist and are in phase I or II trials in kidney cancer could potentially be used against patients against this variant. That could be a very powerful tool, and a kind of the precision medicine that were looking for.
This is an amazing achievement, actually going through the Cancer Genome Atlas to find information like this, information about expression. In the realm of the everyday cancer doctor with patients to treat today, tomorrow, what you think the doctor should take home from this development?
The exciting thing about this is to genotype the patient, that is to determine that this patient carries this risk variant, is something you can detect from the patient’s blood or even a swab from the cheek. It’s a very inexpensive. It costs about $10-$70 to get this information for a patient. You can have what is called a liquid biopsy, meaning you need any tissue. You can get it from their own normal cells, because this is germline variant. You can find out this information very affordably and very quickly to determine risk for these patients. Obviously, if we are able to show that it has implications for therapy, that as a whole opens a whole new avenue.
How much hope to have that this it will be possible to manipulate this gene expression by using this kind of drug to target this?
I think that the data there is quite strong for other types of cancers. We know that other genes that are overexpressed or mutated in activated fashion respond quite well to inhibitors. This exists in multiple cancer types, lung cancer, breast cancer, for example. It stands to reason that this would work as well in kidney cancer, and the hope would be that this variant would be actually an activating factor and that we could use that also.
We’re also hearing, and especially at this GU meeting here in San Francisco, about the heterogeneity of the tumors. In fact, you may have tracked down one particular cause of cancer, but there’s another five going to rear their ugly heads at the same time. What you make of that?
Well, that’s definitely a major factor, particularly in kidney cancer, where heterogeneity was really first described in the clinical setting two years ago in the New England Journal. What the nice thing is about this variant is, is that it is germline. It exists in every cell. Thus heterogeneity does not exist in this situation. The variant is present in all cells, including the tumor cells. So if our data does hold up, and it is a therapeutic target, it will not be affected by tumor heterogeneity.
Give me a message to take home for the community cancer doctor very briefly..
The messages that we have truly validated this important finding that was published last year and we truly believe that this is a new prognostic marker and adds to the existing prognostic markers. Time will tell if it will actually help guide treatment of metastatic disease and really change the paradigm for kidney cancer.
This is a call to sign and save research data that may disappear and not be used for any purposes, which could impact not only current patients, but others who may be affected by this research. We as cancer patients are aware of the value of other research suddenly being valuable to us. Having been diagnosed with a potentially fatal aneurysm (all OK now), I jumped at the chance to help. Please do sign onto this petition. The link is below.
After 10 years of gathering data and tissue samples, this ongoing study has been canceled. All the work will simply be lost. The study is the major hope for people with potentially deadly connective tissue syndromes including fibromuscular dysplasia, Ehlers-Danlos syndrome, aneurysms, Marfans, and Stickler syndrome. We have communities at Smart Patients for FMD and EDS, but they don’t have enough people to carry this petition. Only a few hundred more signatures are needed. Help keep hope alive. Please sign the petition. Please share this petition with your own community and your friends elsewhere, just as been done at my favorite site, https://www.smartpatients.com
This young woman–aged 15–has just ‘explained’ in clear and articulate terms why it is so hard to get both well and respect in a hospital. Must it be this way? Though it is referred to as a “rant”, that is not really the case–it is an indictment of the lack of respect for patients found too often in the health system.
Morgan has some serious health challenges, and has had to learn to advocate for herself to work to get better. But the concerns she raises are typical for most patients, and the lack of coordination of care, and the lack of communication of care is huge barrier to getting well. As do most of us, she understands that the failure to let her sleep is a barrier to that care. And the further failure to let her wake up and be a part of her own care, communication symptoms, asking questions, is far too common.
I congratulate Morgan and her family for sharing this very important reminder.
One of the warnings about kidney cancer is that it is sneaky. Researchers call it aggressive and insidious in nature, as there is a 20-40% recurrence rate for clinically localized disease, i.e, small, hasn’t spread, not to worry, etc. There are many patients who feel reassured by the doctor, generally a surgeon, that “we got it all”, and that there was no need for additional follow up. No CT scans, no blood tests, no nothing.
Most patients are pretty grateful until RCC lives up to its sneaky reputation and makes a surprise return. Since the return is indeed sneaky, is it also sporadic? Is there a way to know which of those patients might need far closer monitoring, or should all of these patients have multiple CT scans or just wait until there is a return. Most early tumors are found incidentally, while checking on something else. That “lucky” patient with a RCC diagnosis may be part of the group which will never have another problem again, or part of the 20-40% who gets “lucky” again. That return of disease can also be silent, with the patient at an advanced stage and in far worse shape than the first time around. What to do? CT scans have their disadvantages, and living under a cloud is pretty hard, and getting RCC again beyond discouraging.
Those nameless researchers, for whom I say prayers of thanks often, have a new tool to determine which early RCC tumors are naturally more aggressive. With this info, patients can be monitored more closely, while the others can live with greater confidence. We’ve been hearing about BRCA genes in breast cancer, thanks to the attention-getting Angelina Jolie. Now we are learning about a related protein in RCC. The expression of this protein helps refine the risks of the early stage RCC patient.
Now it gets a bit technical,but it is important to understand the science here to understand its impact. he expression or lack of expression of some genes can impact prognosis, or clinical expectations, in cancer patients. In clear cell RCC, not the rare variants,such as papillary or others, the lowered or negative (or lack of) expression of BAP1 may signal a cancer that is naturally more aggressive than others. BAP1, also called BRCA1 associated protein-1, is an enzyme which plays a role in cell development, can be mutated or changed in breast and lung cancers, which has been recognized for some time. Recently the Mayo Clinic released a report which indicates that the lack of BAP1 in early stage RCC was associated with greater risk of death for those patients. This is important stuff.
How do they know this? The researchers can detect that expression in tumors. They compared its presence with the outcomes of patients described above. They used 1,479 tumors from patients with nephrectomies for localized ccRCC. This is a very large sample, something important in any trial or research of this nature. They were able to test 98% of the samples provided, and found 10.5% were negative for BAP1, 84.8% were BAP1 positive, and the balance were unclear. After 8.3 years of following patients (Notice how long it can take to get GOOD data.), 1,092 patients were alive, and 252 had ccRCC specific death. Those patients who had BAP1-negative tumors were at a threefold increase risk of death compared to those with BAP1-positive tumors.
Thus, the researcher advocate using BAP1 staining, or analyses, post surgery, to monitor those patients at greater risk of recurrence and death from this subset of ccRCC patients who are likely at greater risk.
All the nagging that kidney cancer patients do to one another to be monitored, despite having had small tumor which was supposedly completely excised is not as effective as it should be. Neither is the “Don’t worry, we got it all” attitude that too often impedes a proper monitoring. This new tool is more objective and should be part of the post ccRCC surgery monitoring.
Just to stir up extra trouble, there may be a case for getting a biopsy to use for this testing, when the small, incidentally found tumor is “slated to be ablated”. Would a biopsy be appropriate, in order to see the level of this protein and the aggressiveness of the tumor? Stay tuned.
Is kidney cancer really so resistant to radiation? Many studies indicate that kidney cancer is far more resistant to radiation therapy than many other cancers. RCC just doesn’t behave as other cancers do, so the most knowledgeable doctors use radiation very carefully. The typical wider damage that comes with general radiation is not balanced by a good response in RCC. To be effective, a more specialized radiation is needed.
One of my most knowledge SmartPatients/friend has provided important information about radiation and RCC. The following is a link to videos, and is followed by advice to a fellow patient trying to understand her options. SBRT is Stereotactic Body Radiation Therapy, and not at all the same as general radiation, as is explained..
“For right now let me sort of define whats important about SBRT and why it works on RCC. SBRT is all about high daily dose needed for radioresistant cancers.
What is important is the high daily dose for RCC used in SBRT. For example, an SBRT plan for a lung met will use 3 fractions x 15 Gy/day or 5 fractions x 10 Gy/day. Fields will be very conformal to the tumor (but usually not by using shaped fields)
Forget shaped. Conformal small fields–yes.
An IMRT (Intensity-modulated radiation therapy) or shaped conventional plan might use 25 fractions x 10 Gy/day.
And to give daily doses of 15 Gy you need excellent imaging and mechanical capability that not every machine has.
Does it matter? Yes! The recurrence rate for RCC is much higher with low daily doses. The SBRT dose regimes will give about 90% local control (meaning that met never comes back-if you get cancer, it is a new spot.) Standard dose regimes give more like 60% long term local control.
SBRT typically uses dose modulation(which may or may not involve shaping beams ) to control where dose goes.
MLC or block shaped beam IMRT IMRT/IGRT-all use shaped beams but are not SBRT.
Cyberknife and Gammaknife, for example, use no beam shaping-they use hundreds of tiny identical circular pencil beams to build dose covering the tumor.
ANALOGY: Let’s say you have a huge thistle in your lawn. And you have a cup of Roundup. You can go outside 3 days in a row and put 1/3 of a cup on it. Or you can go out every other day for a month and put a spoonful on it. You are more likely to kill it for good with the first method. We want a quick thorough cell kill.”
This SmartPatient does not mince words. We all want a quick through cell kill, which may bring a met under control permanently. Not mentioned in this article, is that the body can also have an immune response in the area of the radiation, and deliver a burst of its own cancer-fighting proteins to the area. This can have an additional effect in countering those super-tiny mets that may be invisible, or just trying to establish themselves in the neighborhood.
Just as there are a range of antibiotics, and we know that the choice of each is dependent up the infection, its location, and the individual’s system, so it is with radiation therapy. Properly chosen, delivered correctly and to the exact place, with an understanding of the person’s disease, it may be a very effective tool to fight RCC, and not just clear cell.
Why should you care about genomic research? Simple; it could save your life! Want to know EXACTLY which type of cancer you have, and how to choose the best treatment? New hope comes from this research which really examines the nature of the cells that make up your cancer. Pretty important stuff.
Genomic research is bringing improvements to care, and points up the need to be aware of this new knowledge–and if your own doctor is keeping up with that type of data. At a 2012 Conference sponsored by the The Oncology Journal, Dr. Kimrym Rathmell spoke in regard the genomic knowledge that is leading to improved care for kidney cancer patients. Maybe the most critical lecture of late.
Dr. Rathmell begins after introductory remarks; Complete access on YouTube via this link:
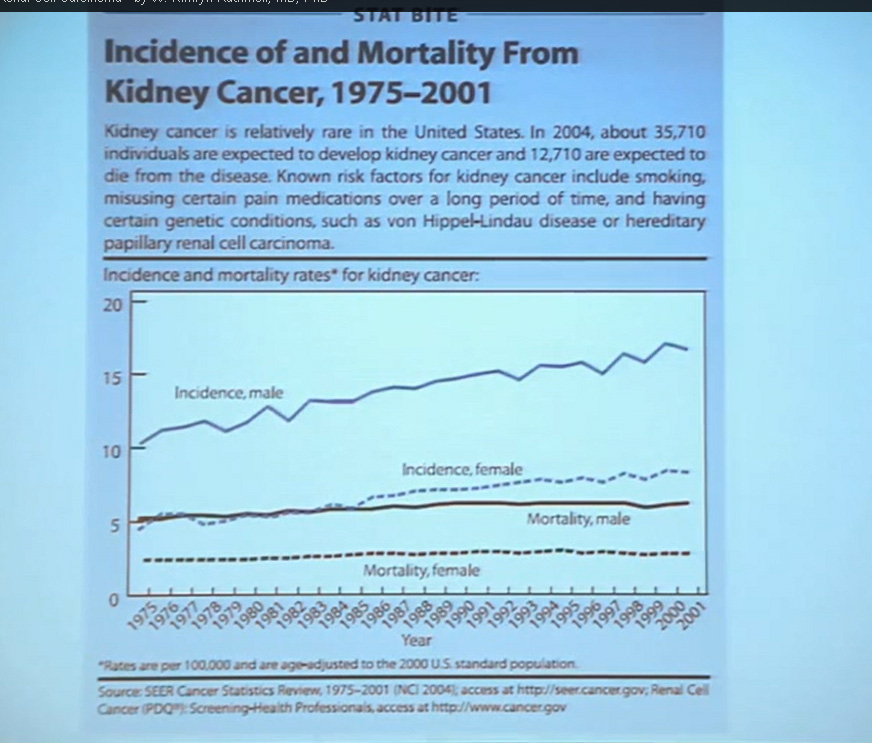
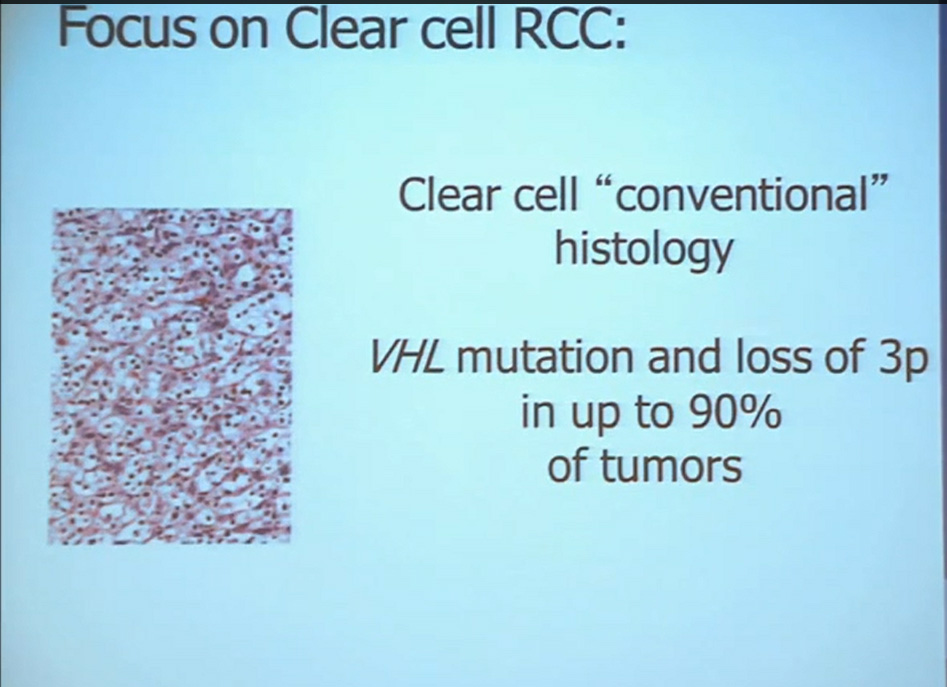
First, kidney cancer, like pancreatic cancer, has been on the rise. This is a somewhat dated slide, dating back to the 70s. We have seen a steady increase in this cancer. Although it was originally characterized as a rare tumor type, it is not really anymore.This talk will focus on one subtype of kidney cancer, that is, clear cell histology renal cell carcinoma. This is a histology slide showing why it is called clear cytoplasm
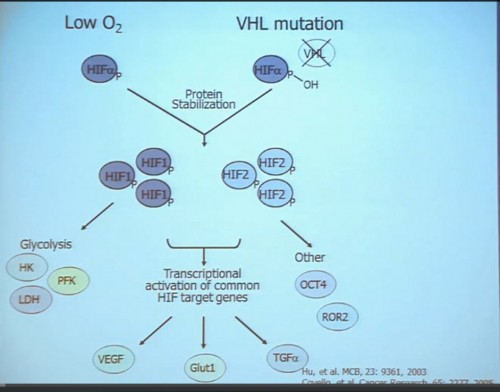
This tumor is characterized by particular mutation. That is the Von Hippel Landau gene, coordinate the loss of 3p, (a chromosome) where VHL is housed. We see these in mutations and loss of 3p which house other tumor suppressors as well, in up to 90% of these tumors. Based on this strong correlation between clear cell renal cell carcinoma and the VHL mutation, a tumor type. is a very distinct paradigm in which VHL loss causes upregulation of hypoxia inducible factors (HIF). These tumors are characterized by loss of high loss of these HIF factors. These are transcription factors that normally allow cells to respond to low levels of oxygen by turning on a repertoire of genes that allow them to bring in new blood vessels, to shift their metabolic properties, to migrate away, to promote survival, and to de-differentiate. That is a perfect storm for kidney cancer, in some respects.
Targeted Agents in Use
Because this cancer has highly nonresponsive to typical chemotherapy, there’s much effort in recent years to develop targeted agents. These targeted agents to date all focus on this well-known pathway in clear cell type renal cell carcinoma. Most of the agents focus far down on this pathway, including that of receptors of VEGF and PDGF. They are tyrosine kinase inhibitors, effective at reducing the tumor angiogenic profile and can be quite effective at reducing the bulk of these diseases. Other drugs similarly target this pathway, for example, targeting features of the tumor that enable HIF to be stabilized such as that in the mTOR pathways. Temsirolimus and Everolimus are approved for use. There are in-developments drugs for targeting MET, which is another mutation that can occur in this cancer, similarly increases HIF levels.
But the reality of treating kidney cancer is that the available drugs that we have do not produce complete responses. We only work in the arena of minimal response and partial response. The extent of response that a patient gets is unpredictable. The duration is also unpredictable and the toxicity is also unpredictable. For drugs we expect them to be effective on average 1 to 2 years, this is chronic therapy, very expensive, and it’s dominated by effects that are substantially detrimental to quality-of-life—fatigue, rash, diarrhea, as well as laboratory abnormalities that indicate damage to the liver or elevations of glucose and cholesterol.
PART I: Clear Cell Renal Cell Carcinoma, Molecular and Genetic Contributions to
INTER–Tumoral Heterogeneity.
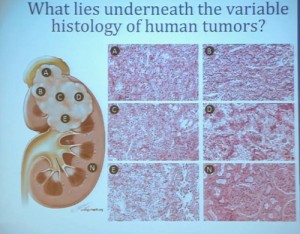
With that, I will talk about various molecular probes that we use to understand some of the diversity or the heterogeneity of these tumors across the clear cell renal cell carcinoma spectrum. Before I really dive into clear cell renal cell carcinoma, I need to point out that there are other histologies with this tumor as well. So when we say kidney cancer, we’re talking about a big spectrum. Clear cell renal cell carcinoma, we are talking about those tied to Von Hippel Lindau disease and loss of 3p and it is about 70% of all cases we encounter in cancers of the kidney. But there are also other types. Papillary type renal carcinoma, chromophobe, benign tumor—oncocytoma, a translocation form and some very rare. With these types of tumors we have very little in terms of knowledge of how to treat these patients. Their genetics are highly distinct from clear cell renal cell carcinoma. So someday in the future, we will understand not only how to treat not only our clear cell carcinoma patients, but how to use effective molecular information to target these cancers as well.
Clear cell carcinoma is well know to be molecularly heterogeneous for some time. This is a gene expression profile. We’ve already seen heat maps from several of these other talks, looking at gene expression profiles. And as you can see the gene expression profiles across a large selection of tumors here, suggests there are great areas of variability–at least two and as many as five groups, based upon gene expression purely.
Pattern Recognition Profile to Find Subtypes
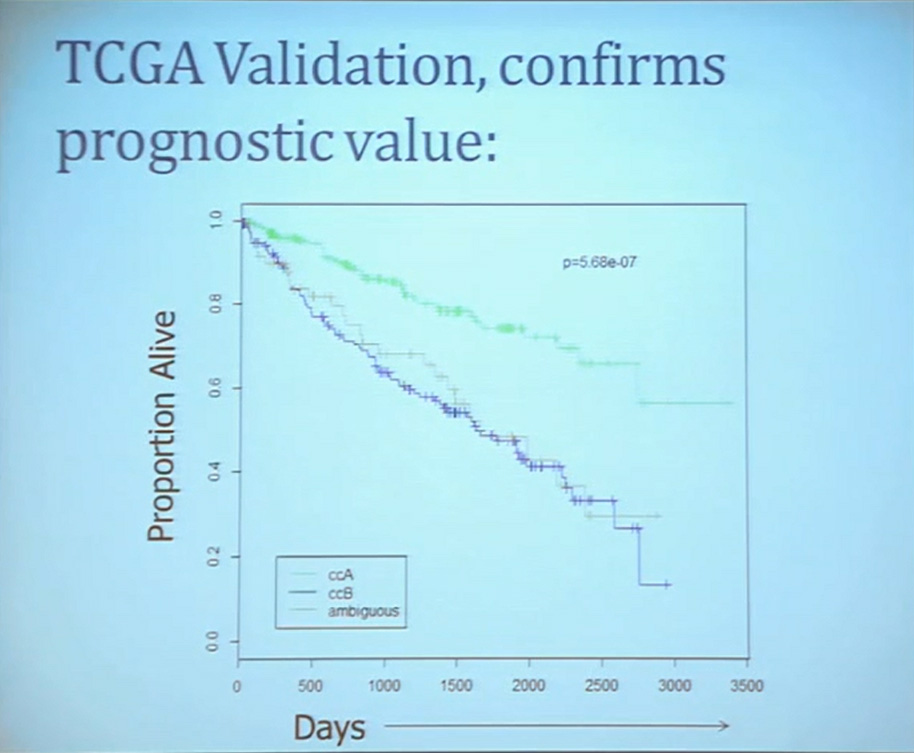
Our group undertook at the time developing a pattern recognition profile which is now fairly routine use. To try to see with a more robust computational strategy what subtypes we could really identify, that we could really pen down and understand with genetic profiles. 8aWe found two. For lack of better knowledge, we are calling clear cell A and clear cell B, ccA and ccB. These are very distinct biologically, and when we look at these tumors in terms of their outcome, they also have significant prognostic relevance–with the ccA tumors in this original cohort having a median survival of 103 months, compared to the 24 months for ccB tumors.
The TCGA which is been discussed here in many of the previous talks is a great source of validation. We assigned these tumors to clear cell A and cc B groups subtypes, validating our previous results with the clear cell A tumors having much better survival profile than those ccB tumors.
This classification scheme, which is based 120-130 gene signatures classified robust subdivisions of clear cell type renal cell can be applied with a small number of genes on individual tumors and is independently associated disease-specific overall survival, making it a valuable prognostic biomarker.
PART II: Rare Variant Groups
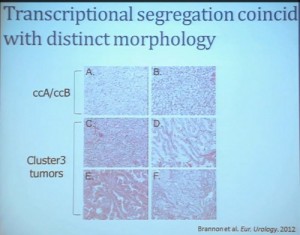
We use these profile tools to understand the rare variants. This is still in the clear cell renal cell carcinoma arena, but when we took a very large group of compiled tumors; this was a meta-analysis of 500 tumors, all histologically defined as clear cell type renal cell carcinoma, and we applied our expression pattern recognition algorithm. We asked for two groups and we found two and they correlated with our ccA and ccB, but when we ask for three groups, we can find a small group that now filters out. Now that we have power in numbers to identify what we called Cluster 3. What is in Cluster 3?
Cluster 3, as we’ve said, is histologically defined as clear cell renal cell carcinomas. But we look to the genetic expression profiles, it’s very different, particularly with regard to metabolic properties. We see upregulation of genes that are involved in mitochondrial regulation and oxidative phosphorylation, suggesting a striking difference in the way these tumors likely regulate metabolism.
In addition, and now these are tumors that we can not go back and genotype for VHL mutation, for loss of chromosome 3p, but the loss VHL regulation leads to characteristic changes in the gene expression profile. So when we use the gene expression changes to predict whether these tumors have an intact VHL or a mutant type VHL. The wild type VHLS signature shown here is shown in purple. You can see that these purple tumors, the wild type VHL tumors all tightly cluster with Cluster 3. These are probably not clear cell renal cell carcinomas although many, pathologists call them that. So we pulled them all out so, asking, “DO they look a little bit different?” My graduate student, who did this work, came right away and said “There’s something funky about these clear cell tumors that we call Cluster 3.”
As you can see, these are clear cell A and a clear cell B tumors, but they all have the clear cytoplasm and really, what we are seeing, is that they are not distinct histologically, although they are very different molecularly. And as I have shown, they have a very different prognostic outcome. The Cluster 3 tumors; although the cells themselves might have clear cell cytoplasm that gave them the clear cell histology designation, they have a very different pattern of organization—with a papillary type of feature. So what we think it would be identified as is a new rare variant of clear cell renal cell carcinoma.
Simultaneously another group of pathologists identified, that the pathologists call clear cell papillary carcinoma. That suggests that we need to take a very great care as we treat these patients. What we have is clear cell type renal cell carcinoma, most of which are VHL-mutated, and we do have clear cell A and clear cell B. These are the tumors we should be treating with the drugs with identified, based on the effect of the pathway that is activated by loss of VHL. But clear cell papillary renal cell carcinomas probably won’t react very well, as they are VHL-wild type. Just like papillary renal cell carcinomas don’t react well either.
To summarize this section, clear cell renal carcinoma can be separated into ccA and ccB groups, based on transcript profiling, but further clustering can identify highly biologically dissimilar subtypes within the clear cell group, and that subtyping can convey a biological distinction which is a valuable tool for prognostic evaluation, and a likely cause of poor responses to some therapy.
Part III; Using Clinical Trials to Understand Biological Relationships to Response to Therapy
As my title indicated, we also refer to clinical trials to help us understand renal cell carcinoma a bit more. A clinical trial we completed some years ago, LCCC0603, was in neoadjuvant trial that looked at the treatment of renal tumors with sorafenib. Patients were identified as having renal tumor and underwent CT scans for basic size, description and PET scan, and then were treated with sorafenib. It is the first generation VEGF receptor tyrosine kinase inhibitor for 4 to 8 weeks, and then underwent post treatment CT scan, PET scan and a nephrectomy. We are going to look at radiographic indicators of response, rather than molecular indicators.
Waterfall Graph with Response at 30% as goal
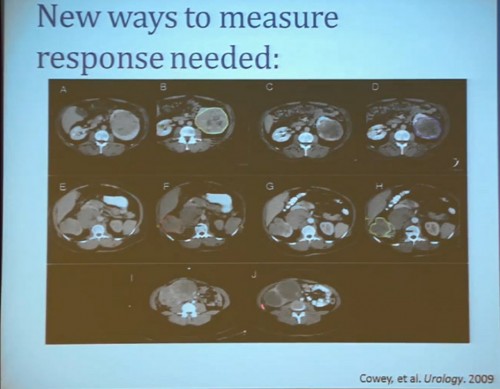
First standard RECIST criteria do show that we do see partial responses. Again, there were no complete responses. Many had subpartial or minimal or some partial responses. Their tumors shrank, but most did not meet the standard criteria of 30% decrease in one longest size. Some tumors actually grew.
Now what we realized as we looked at these tumors, is that we probably need new ways to describe response. The standard RECIST criterion response is just based on longest diameter and measuring this in comparison, after treatment..
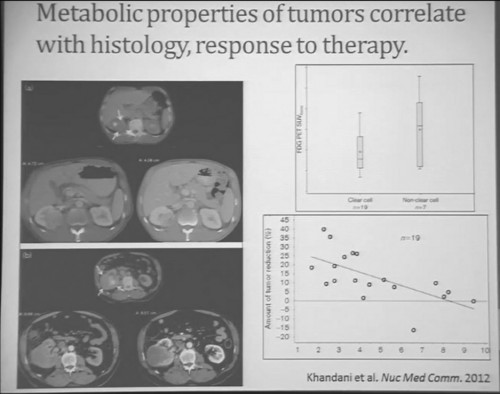
I will use this patient is an example. (References I and J, in the lowest row). Here is a pretreatment; we have a very large renal tumor. Post treatment, the tumor was still large, but it measures slightly larger than it had been before. But if you look at this tumor, it is very different. The central area of this tumor is now very dark, indicating necrosis—is what we think. But we took these tumors out, we could confirm that these dark areas were indeed necrotic. So we developed a new way to try to quantify the area of the tumor that is actually killed in response with this treatment.
Similarly we were doing PET scans on these patients, and we were doing this because we’re trying to understand how the metabolic properties of these tumors might indicate how these tumors were likely to respond to this treatment. We see, and have known, that are some tumors which are very dim on FTG PET. This is a tumor; (Smallest of upper images) you can see that here is very visible on the PET scan. It doesn’t take up any FTG. So this tumor has the metabolic profile that is not dependent upon uptake of glucose. Others; this tumor (Smallest of lower picture), for example, have regional areas that are can be very high in terms of FTG uptake. When we looked at these tumors, we discovered first that non-clear histology tumors were much more likely to have high levels of FTG uptake. So, metabolically active tumors more likely in the (correction) non-clear cell group, probably the papillary, the chromophobes and the papillary clear cell types, than the clear cell group. Secondly. we discovered that the correlation between FTG uptake and response looked somewhat different than we might have expected. We might have expected the most metabolically active tumors would be those that would response better to anti-angiogenic agent. But the opposite was true. The best are those that had very low levels of uptake FTG uptake. We are still trying to understand exactly what that means. Certainly that means that those clear cell tumors are the ones more like to respond, activity, what we have known, but those are the ones with the lower level FTG activity. But we continue to try the metabolic properties of the tumors that make theme different more likely to respond.
That leads to our next clinical trial. This is now ongoing. This is LCCC1028. It is a neoadjuvent clinical trial using the newest generation—well, they are coming out so fast that it’s the not the newest, but the next to newest VEGF receptor tyrosine kinase inhibitor. They are now getting PET scans and a biopsy to confirm in fact that they would be clear cell renal cell carcinoma, and also to allow us to do molecular studies that directly measure their metabolic activity and other effects. They’re being treated for eight weeks with another CT scan, undergoing nephrectomy. We will then be able to look at clear cell variant histology. They will all be clear cell going in, but there may be variants included–as well as looking at their VHL mutation and their other mutational status, their transcript profile, in particular the clear cell A and clear cell B group and other protein expression signatures.
This, of course, is known for all tumors, but if you sample in multiple different places, the histology will look different and in effect, the grade can look different depending on where you are sample.
What does that mean molecularly? Well, a group at the Sanger Center published on a small number of tumors. When they sequenced these tumors, they found while there are some mutations that are ubiquitous, meaning the mutation is found in all samples across the primary tumor and the metastatic tumors, that there are mutations that are private. There are mutations that are common only among the primary tumors and there are mutations that are common only in the metastases, and there are a lot mutations that are unique to the individual sample. This makes a whole new level of complication as we moved toward personalized therapy, in particular therapy that is based on biopsy metrics.
This group also looked at our clear cell A and B subtypes. And what they saw, when they looked at six samples from the primary tumors, was that in five of those samples, the gene signature indicated that these would be clear cell B type tumors. So depending on your glass half-full/glass half empty: The glass half full version of this, that five out of six times, they would pick that the patient would have poor outcomes. This patient has metastatic disease, so it fact, that is true. The glass half empty would be that one out of six times, he would pick wrong. This patient would have been indicated to have clear cell type A tumor, and you might have predicted that this patient would do well, when in fact that would be wrong. So what helps us understand the limitation of this test. It also gives us the opportunity to understand a little bit more about these tumors.
(Footnote reads: BRIC Funded Grants LCCC1213) So for the future, a trial (LCCC1213) that we have really just initiated is uniting some of these imaging observations we have made with genetics.
We are taking in patients. This is patient number one. Patient number two just has his MRI last week. and doing an MR in coordination with the PET scan so we can get the detailed look at these patients’ tissue perfusions, vascularity and the density of these tumors, as well as regional areas at the FTG uptake and sample according to the map that is created by the imaging, as well as samples that are collected, based on what we see grossly in the tumor. Here you can see a sample that we collected from a tumor region that is highly distinct from the mostly more pale yellow regions of tumor.
This is just begun, so I cannot tell how well were going to get to correlate the gene expression and genetic underpinnings, and what we see in the tumor and what we see in the MR / PET. But it will help us to move forward.
To summarize are multiple ways for RCC to diverge. The subsets can enrich tumor sets for clinical and genetic features, and a multiplatform approach that with genetics, molecular biology and imaging techniques will give us man ways to tackle a surprisingly very heterogeneous disease.
Once a patient can stop asking why cancer happened to him/her, the next question is the fundamental version of the many questions that the researchers ask– “What is cancer really? and “Why can’t they just stop it?”.
The scary part of cancer is that it seems so insidious. Cut it out. Burn it out. Zap or freeze it out. Why doesn’t that work, at least for the solid tumors? Getting rid of the blood cancers, like leukemia and lymphomas seem more difficult, less obvious, but it was really the cases of leukemia which first responded to treatments.
Without going into volumes of discussion about cell and molecular biology (you are safe from that with me), just understand that cells go wild, left to their own with the family checkbook, an endless liquor cabinet, permanent pizza delivery, car keys, disguises, blind neighbors, a fancy cloning machine, and the police on strike. You get the picture. Now a more formal explanation.
Cells are supposed to do their respective duties and then die. That process is called apoptosis. You know that your scabs don’t keeping growing, but cancer cells lose the “time to die” signal.
Foreign bodies are supposed to be cleared by the immune system, and what is more foreign that cancer? However, cancer cells manage to evade immune destruction. And while doing that, they can also evade the growth suppressors, the immune brakes which would otherwise slow and prevent excess growth.
While cutting the brake lines to growth, they can also change the regulating signals for growth (think scars and healing), so those signals all left in permanent “ON” position. No brakes and an open throttle with a very full tank of fuel. To top that, they reprogram the nutrition or energy metabolism to keep the fuel of growth alive.
Liking the growth, the cancer cells override signals that naturally limit the times a cell can divide, creating endless replication instead. With the endless replication, the chances for mistakes, or genetic mutations increase, which can mean changes from the original cancer cells. Sheesh, not only alien cells, but aliens cells making alien-er cells!
To keep tumors growing, cancer cells send out signals to create blood vessels or angiogenesis when tumors outgrowth the local nutrient sources. Running out of room for all these many cells, billions and billions, the cells break down the lining of blood vessels and the lymphatic system to search out new locations, spreading and metastasizing away from the original cancer. Quite naturally, they also provide support to those tumors through inflammation-related factors, mimicking the way that the immune system responds to any injury.
Since a healthy being grows, fights off infections, responds to an allergy or heals after an injury, usually with little support, those healthy responses are amazingly efficient and interactive. Complex cellular, molecular and chemical actions are occurring all the time, and with aging, some genetic dispositions, the harmful things we ingest or do to ourselves, it is no wonder that a few things go on without our noticing it. But when those few things are not noticed by the immune system, slipping into a growth phase, a cancer can begin.
Estimates of the numbers of cells in the human body are calculated from 10 trillion to 100 trillion, so if the occasional cell goes rogue, what’s the problem? When all things are working well, there will be no problem. But when the tiniest cancer is visible with a CT scan, perhaps 1/8 inch, it will have millions of cells. Not all tiny cancer tumors are dangerous. Not all become aggressive. Digging around to cut out a tiny tumor creates plenty of opportunity for infection, for expense and emotional anguish. But does that mean that a “Cancer!” has been prevented. Or would the person and his natural immune system have lived in complete tranquility with his cancer until the end of his days?



















 QED
QED