Recent headlines called a new medication, Nivolumab, both a miracle or breakthrough and more. Is it hype or hope?Why is it so hard to sort out the reality?
Let’s go through the facts from the New England Journal of Medicine and ignore the headlines. First, its being in the NEJM is important, as it has passed review by other researchers. (Sadly missing in too many ‘breakthroughs”).
The new med, Nivolumab was compared against Everolimus, a second-line treatment. Therefore Evero is thought to be of lesser effectiveness than the first line meds. Second-line meds are generally used when others meds quit helping or their side effects are too hard. Automatically NOT the miracle cure, but another option when first-line treatments fail.
Should Nivo have been compared to the first-line meds? Being better in the first-line would be bigger deal, but we need more approved meds. Second-line treatments usually are easier to ‘beat’, as the new med must be better or less toxic. Again, more likely to be approved!
PATIENT CHARACTERISTICS
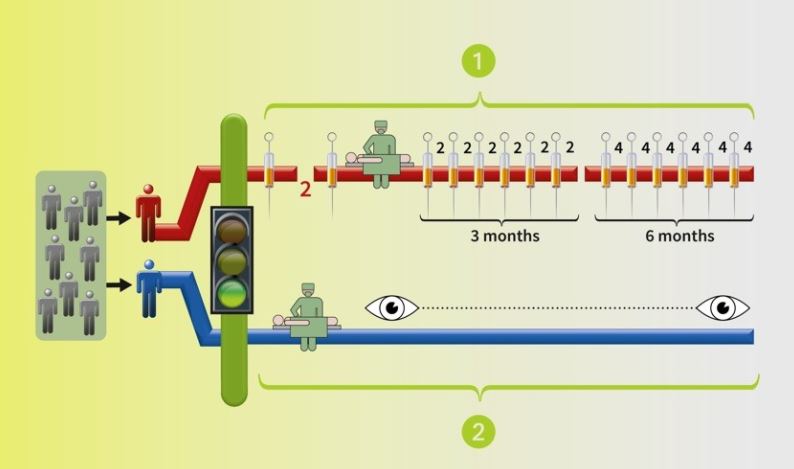
The study had 821 patients 24 countries, half using Nivo and half Evero. Patients were similar, 90% having had a nephrectomy, removing the tumor and some or all the kidney. Then the cancer spread, making metastases, (mets, for short). These patients had 1-3 treatments, first-line drugs like Sutent, a targeted therapy, and a few had used cytokines or even chemotherapy. Having had an mTOR inhibitor like Everolimus was not acceptable. Most had lung mets (67%), followed by liver(12%) and then bone mets (18%). Most with 2 or more sites of mets.
To enter the trial, the patient had to have had disease progression after their last treatment, within six months of enrolling in the trial. No doubt, some patients had greater disease progression than others, but had relatively good performance status, not completely bed-ridden or unable to function.
The median time from initial diagnosis of kidney cancer at any stage to entering the trial was 31 months; half had been diagnosed less than 31 months ago, and half more than 31 months before the trial. That range of time from diagnosis to trial was 1 to 392 months. That means that for some patients, they went a long time either fighting the disease since diagnosis, having a later recurrence, being treated, yet having disease progression years after the intial diagnosis. At least one person was diagnosed 392 months earlier. This is a good reminder to patients who have been told, “I got it all”. This darn stuff can return, so having a plan B is important. Again, the previous treatment failed and these patients got directed into this trial.
GENERAL RESULTS
Median Overall Survival (OS) is a measured when one-half of the total number in the group dies. Median OS for Nivo was 25 months with some patients still surviving at time of report, beyond the 25 months. For Evero, OS was 19.6 months, some of who were also likely surviving, as well. The OS of 25 months was clearly better with the Nivo group by this analysis. Nevertheless, half of all the 821 patients total died while on this trial from progressive disease. Of 183 of the 410 Nivo patients, 183 has died by 25 months, and 215 of the 411 Evero patients had died at 19.6 months.
There is no report of ongoing response here, but many went on to other meds, as explained below.
Median Progression Free Survival (PFS), measurable growth of disease, was 4.6 months for Nivo, 4.4 months for Evero. The median shows that half of each group, roughly 200 each had return of disease in less than 5 months! Again, these trial patients were pretty sick or at risk. All had been treated earlier, and had to stop previous treatments due to recurrence of disease. However, this shows a pretty quick return of disease or new growth from the base CT scan for nearly 65-70% of all patients.
One subgroup did a bit better than the 4 1/2 months median PFS. At six months after the start of treatment, there was a special subgroup was noted, about 1/3 of those patients–145 pts (35%) with Nivo, and 129 (31%) with Evero. Obviously they did not die or have Progressive Disease until after six months. The Nivo group had eventually had a median PFS of 15.6months, and the Evero group, 11.7 months. Their success pushed the median OS higher, especially for the Nivo group.
Obviously, there were some patients with far more aggressive disease in both groups, some dying before six months, and others not progressing to more disease until after six months. In contrast, nearly 1/3 of all the patients had PFS of 12-15 months, and much longer OS. What is the common characteristic in the most successful two groups in both arms of treatment? Not answered by this trial report.
The duration of treatment was longer with Nivo, and likely easier to tolerate. Since Nivo was given by IV every two weeks, the doses were most consistently received. Even so, 51% of them had dose delays, but no per dose reductions. Those people were seen by the medical team every two weeks.
The Evero group took oral meds, and 66% had dose delays or interruptions with 26% with at least one dose reduction. This would indicate that these meds could be hard to take, or perhaps lacking the same interaction with their medical team. Of course the Evero patients may have underreported how much of the medication they actually took!
However, the reported types of side effects were generally similar, but the more severe grade 3 and 4s effects in the Evero group.There were 2 treatment related deaths in the Ever group, none in the Nivo group.
POST PROGRESSIVE DISEASE
Even after the disease did progress, about half of patients in both groups stayed with their meds–despite ‘failing’, the researchers hopes that would continue to benefit, perhaps slowing the disease. In a local clinic setting or with a less experienced docs, their meds might have been stopped or changed. Afterall, those meds were no longer “working” and mets are growing. This approach is significant to consider, especially after multiple treatments. (The decision to keep giving a medication or increasing its dosage where tolerable is causing some changes in treatment in a number of the targeted therapies.)
Perhaps because of being in a trial or getting care than was more expert than most, one-half of patients chose to keep on the trial meds. Others crossed over to the med in the other arm or returned to existing non-trial meds. In some countries, there were likely fewer choices than in the US. There are no real stats as to survival for those on those who stopped taking the meds. It is reported that indicate that 55% of the surviving Nivo group and surviving 63% of the Evero group went on to other agents. About one-quarter of the Nivo group shifted to the Evero. Of the Nivo group, 36% shifted to axitinib.
Sadly, as per the chart in the New England Journal of Medicine, all these patients had died by 30-33 months post enrollment. However, it is again not clear what was effect, if any on that period from the non-trial drugs. Of the 227 who stopped Nivo for any reason, nearly half shifted to Evero. Of those who stopped Evero, 140 went to Axitinib.
DURABLE RESPONSES? HOW LONG? FOR HOW MANY?
The writers of the study say that there was a higher number of objective responses with Nivo vs Everolimus, and that many (of the Nivo group) “were durable”. There is no definition of ‘durable’. My question is “What equals durable?”. We patients really want a cure, but are very grateful for anything that pushes the cancer back, slows it, stops in from growing any further. Nevertheless, we do want those responses to last. The clearest reference to durable responses is a note that 32 of the Nivo patients and 6 of the Evero patients had a response that lasted more than 12 months. But in an unexplained statement, the median duration of treatment was just 5.5 months for the Nivo patients, 3.7 for the Evero group. It seems that there was not an extension available, or that the patients moved on to a different treatment or passed away.
CONCLUSIONS AND EDITORIALIZING AGAIN
It seems that Nivo is more helpful for some patients than others in this group previously been treated with other TKIs. This is NOT A SILVER BULLET. There would be greater value to know more about the molecular nature of the tumors of the responding and the non-responding patients. We desperately need to know for whom any of these drugs is likely to be more effective. The headlines that don’t discuss that challenge underserve us, as does the design of the trial that does not elicit the more nuanced, genomic data that could be forthcoming!
We all know that headline claims are more wonderful than the reality. The story of RCC medication development is that of more and more help in a difficult disease, making mixed progress, while the other researchers find out that RCC is really many diseases. Clear cell is probably better defined as being made up of four types, Papillary Type 1 and Type 2 being further divided into three Type 2, then there is chromophobe, clear cell papillary and the really odd versions of RCC. I known this, and so do you. But why don’t the researchers incorporate those definitions and monitor the patients with those various subtypes as they go forward?
Good luck to all of us.