How Does the Pathology Report Help Direct my Treatment Options?
Lecture by Dr. Daniel Luthringer of Cedars Sinai Medical Center of Los Angeles at Kidney Cancer Association meeting December 2013. https://www.youtube.com/watch?v=-6emPs-mc1E (Follow via YouTube)
I have transcribed the lecture edited for readability, included the slides, to make it easier to follow. If you have not seen your own pathology, GET THAT REPORT now. Important to read!
A terrific introduction by Dr. Robert Figlin reminds us of the work of those people we never meet, but who care for us. “One of the people behind the scenes is the pathologist at this and other institutions. Often times the pathologist is in a different part of the hospital evaluating tissue, and helping the clinician figure out what the tissue looks like. It’s become, as Hyung (Dr. Kim) mentioned, time to start to think about personalized approaches to kidney cancer, and the relationship between the pathologist, the surgeon, and the clinician becomes ever more important. Dr. Daniel Luthringer is Professor of Pathology and Section Chief of the Genitourinary Pathology. He will talk to us about how the pathology report and how what he does– is important to then what we decide how to go forward with treatment.”
Dr. Luthringer begins:
“Thank you, Bob, for this introduction and the ability to speak at this conference. I am the guy behind the scenes, at least at this institution responsible for doing the histologic/microscopic analysis of genitourinary malignancies, primarily renal cell carcinomas. (RCC)
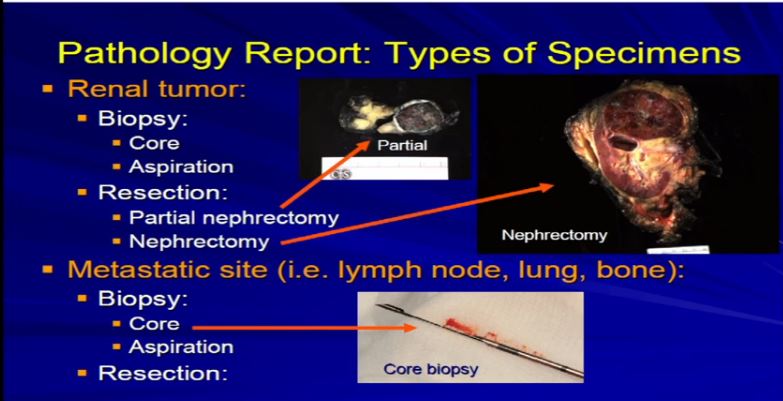
1  There are really two main categories of specimens we receive, samples from the real tumor itself, which can either be biopsies or resections, as Dr. Kim alluded to, or samples from a metastatic site, a recurrence or a metastatic site. The most common specimens that we see are nephrectomies, resections of the tumor, andeither partial nephrectomy or complete nephrectomy.
There are really two main categories of specimens we receive, samples from the real tumor itself, which can either be biopsies or resections, as Dr. Kim alluded to, or samples from a metastatic site, a recurrence or a metastatic site. The most common specimens that we see are nephrectomies, resections of the tumor, andeither partial nephrectomy or complete nephrectomy.
2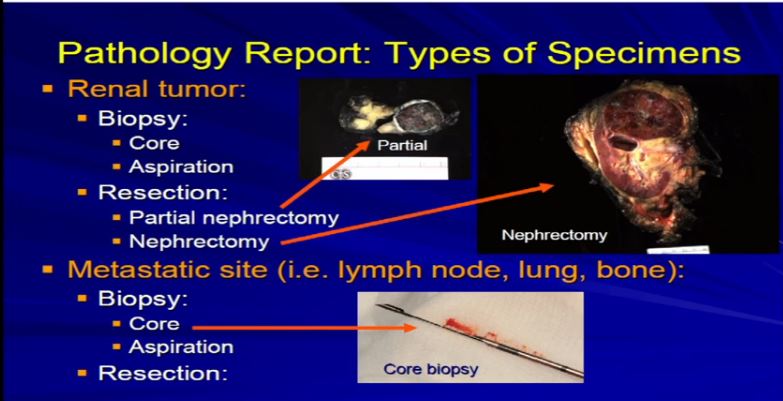 These are examples. A partial nephrectomy, as per Dr. Kim, are smaller resections or partial resections of the entire tumor.They include a bit of nephric fat and a little bit of the perinephretic fat as well. The goal is to get the entire tumor out, with a negative margin of resection. With tumors that are bigger generally, or infiltrative, we tend to get the entire kidney. This is an example of a nephrectomy with perinephric fat, the sinus fat, drainage area down here, maybe an adrenal gland up top and this would be an example of tumor that is completely resected.
These are examples. A partial nephrectomy, as per Dr. Kim, are smaller resections or partial resections of the entire tumor.They include a bit of nephric fat and a little bit of the perinephretic fat as well. The goal is to get the entire tumor out, with a negative margin of resection. With tumors that are bigger generally, or infiltrative, we tend to get the entire kidney. This is an example of a nephrectomy with perinephric fat, the sinus fat, drainage area down here, maybe an adrenal gland up top and this would be an example of tumor that is completely resected.
Occasionally we will get tumors from metastatic sites or—unusually from the primary tumor—and will get a core biopsy, which is really a small smaller sample of the tumor mass. Usually it is about a millimeter or two in diameter; it’s a core, maybe up to several millimeters up to a centimeter in length. Generally, it is just a small sample of a much larger tumor .
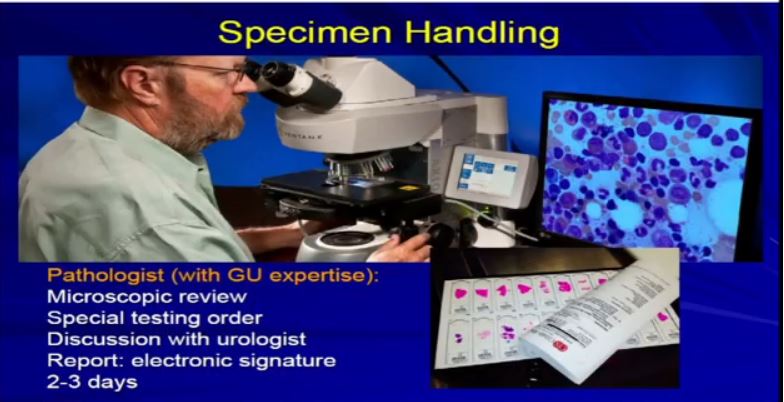
 3 A bit about the specimen handling: within a few minutes of having the tissue removed, it comes to the pathology lab. We do some initial assessment on it. We have work stations where they will come and the pathology team will assess it. Assume it is a nephrology specimen. We look at it and measure it, cut it open, procure some of the tissue. If there is some tissue that needs to be taken fresh, potentially for a biobank to be stored away, or if some tissue needs to be taken for immediate diagnosis or margins or something like that, we will do that.
3 A bit about the specimen handling: within a few minutes of having the tissue removed, it comes to the pathology lab. We do some initial assessment on it. We have work stations where they will come and the pathology team will assess it. Assume it is a nephrology specimen. We look at it and measure it, cut it open, procure some of the tissue. If there is some tissue that needs to be taken fresh, potentially for a biobank to be stored away, or if some tissue needs to be taken for immediate diagnosis or margins or something like that, we will do that.
If you’re enrolled in a study where there some fresh tissue is needed, sent to a particular institution or a reference laboratory for an analysis, we will procure that as well and make arrangements to send it off on an immediate basis. At that point we do photography, tissue fixation and over the next few hours we will dissect the specimen, will analyze it, do a lot important evaluation with our eyes and ears, whatever it takes. Then we will take what are called representative sections of that tumor or specimen, put them. We put them into these little capsules called cassettes and then we process them overnight in these tissue processors. These are pretty standard from institution to institution.
 3a The next morning the tissue is taken out of the processors and is manually placed in these other tissue cassettes which are filled with paraffin wax essentially. They are embedded into these wax molds, and then the blocks. Then very thin sections of 4 to 5 microns are cut with these special microtomes and they are picked up on the glass slides. They are again processed, stained, and cover slipped. Ultimately we get a sample of glass slides from that tumor that has been removed. On an average partial or complete nephrectomy, we will go anywhere from 5-10 paraffin blocks, equating to 5 -10 glass slides.
3a The next morning the tissue is taken out of the processors and is manually placed in these other tissue cassettes which are filled with paraffin wax essentially. They are embedded into these wax molds, and then the blocks. Then very thin sections of 4 to 5 microns are cut with these special microtomes and they are picked up on the glass slides. They are again processed, stained, and cover slipped. Ultimately we get a sample of glass slides from that tumor that has been removed. On an average partial or complete nephrectomy, we will go anywhere from 5-10 paraffin blocks, equating to 5 -10 glass slides.
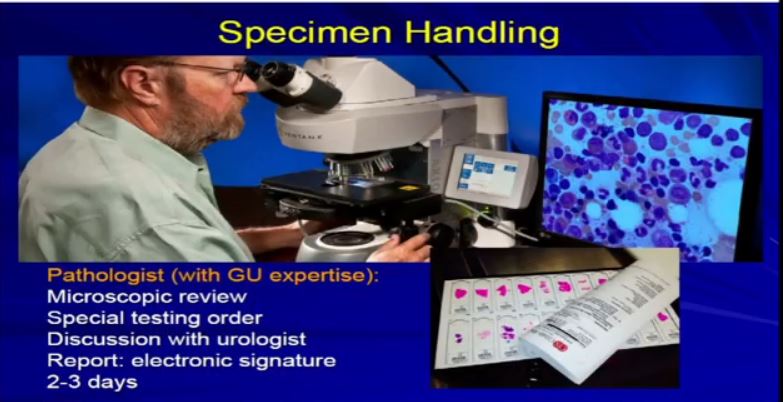 This takes about a day or two to complete this. Then the initial slides are delivered to the pathologist, who will begin the process of microscopic analysis. He uses obviously his microscope and whatever tools he needs.
This takes about a day or two to complete this. Then the initial slides are delivered to the pathologist, who will begin the process of microscopic analysis. He uses obviously his microscope and whatever tools he needs.
He’ll be looking at those sections from the slides, and it will usually be the sections from the kidney, maybe some lymph nodes, margins, adrenal glands, things that were provided by the surgical resection. The whole process usually takes 2-3 days to complete. There is a bit of a time lag, due to the technical processing involved.
 4a The Elements of the Report. Once we generate the report, and it becomes available, there are really three categories of information that are really relevant– not just the diagnosis, but the future care of the patient. The first is the diagnosis. What is the diagnosis? Is it really renal cell carcinoma or is it some other unusual type of renal cell cancer? I will talk more about that. Then: aspects related to cancer stage–tumor size, local infiltration. Has it metastasized or spread? Last, the other features that Dr. Kim alluded to in his talk—resection margins, grade, vascular invasions. We will talk to about these just briefly.
4a The Elements of the Report. Once we generate the report, and it becomes available, there are really three categories of information that are really relevant– not just the diagnosis, but the future care of the patient. The first is the diagnosis. What is the diagnosis? Is it really renal cell carcinoma or is it some other unusual type of renal cell cancer? I will talk more about that. Then: aspects related to cancer stage–tumor size, local infiltration. Has it metastasized or spread? Last, the other features that Dr. Kim alluded to in his talk—resection margins, grade, vascular invasions. We will talk to about these just briefly.
 4bThe first aspect is diagnosis. The important thing to remember, and I think everyone in the room is a little bit beyond this, but remember that at the initial phase, tumors are resected and often times it is not know if it is a RCC. Often times it isn’t even know if it is a neoplasm at all. Not all tumor masses are neoplastic or malignancies.
4bThe first aspect is diagnosis. The important thing to remember, and I think everyone in the room is a little bit beyond this, but remember that at the initial phase, tumors are resected and often times it is not know if it is a RCC. Often times it isn’t even know if it is a neoplasm at all. Not all tumor masses are neoplastic or malignancies.
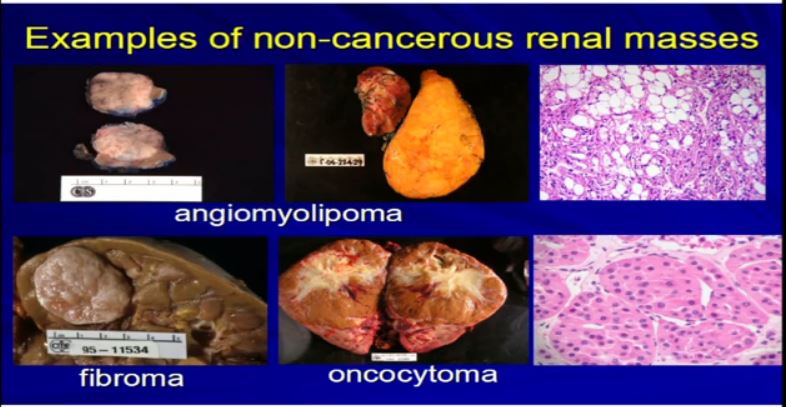
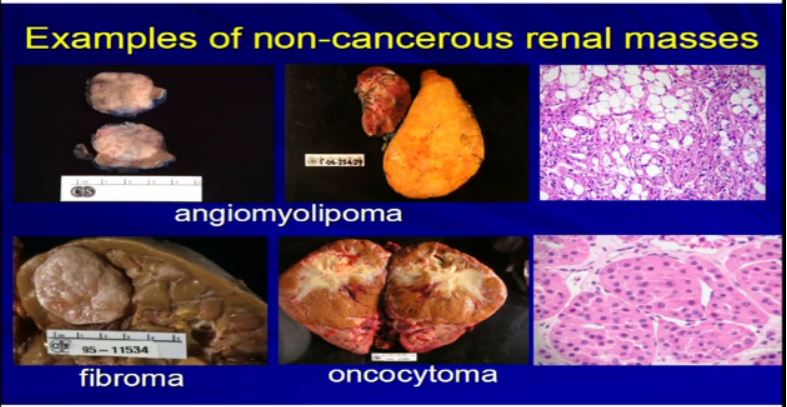 5 Examples of non-tumor masses would be like cysts, a lot of cysts. A lot like this or areas where the collecting system is dilated called hydronephrosis or multiple cysts can present or look just like a RCC. They are resected as if they were RCCs. But in fact they are not—they are benign
5 Examples of non-tumor masses would be like cysts, a lot of cysts. A lot like this or areas where the collecting system is dilated called hydronephrosis or multiple cysts can present or look just like a RCC. They are resected as if they were RCCs. But in fact they are not—they are benign
There are other types of tumors besides kRCCs. Angiomylipomas are a very common tumor. They could be very big like this one. Here’s a kidney. Here’s a big one. They could be multiple. Here’d two. They could be small one or 2 cm like this, but they all look like fatty tumors, but not all RCCs. Different types of tumor like fibroma or oncocytoma can be very big and aggressive-looking, but in fact, they’re not malignant at all.
 6 There are other types of malignancies, true malignancies of the kidney which are not real carcinomas. Urothelial tumors, those that are derived from the lining of the kidney that can extend into the kidney, be derived of the kidney. These are examples of some of these here. They were resected, thinking that these are probably RCCs, but in fact they turned to be urothelial, not RCCs.
6 There are other types of malignancies, true malignancies of the kidney which are not real carcinomas. Urothelial tumors, those that are derived from the lining of the kidney that can extend into the kidney, be derived of the kidney. These are examples of some of these here. They were resected, thinking that these are probably RCCs, but in fact they turned to be urothelial, not RCCs.
Different types of tumors like sarcoma can be derived of the kidney or around the kidney. Other types of tumors can metastasize to the kidney or near the kidney. Adrenal tumors, lymphomas—there is a whole host of malignancies which can mimic RCC.
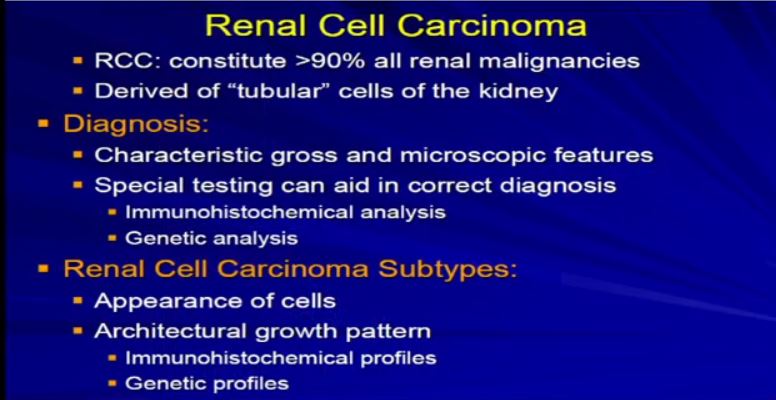
 7 What were really talking about today here obviously is renal cell carcinomas which represent probably 90% of more of all true malignancies of the kidney. These are the tumors which are derived from the renal tubular epithelian cells, those little ducts that line the epithelium of the kidney. The diagnosis of RCC really is contingent upon microscopic analysis. You can’t make the diagnosis any other way.
7 What were really talking about today here obviously is renal cell carcinomas which represent probably 90% of more of all true malignancies of the kidney. These are the tumors which are derived from the renal tubular epithelian cells, those little ducts that line the epithelium of the kidney. The diagnosis of RCC really is contingent upon microscopic analysis. You can’t make the diagnosis any other way.
The pathologist needs to look at the gross, take a section, look under the microscopic, and then there’s a spectrum, a range of features that will ensure the diagnosis or put it into a diagnostic category of RCC. Sometimes is not so simple. We need special testing–the use of antibodies, immunohistochemical studies or even as Dr. Young Kim alluded to, sometimes we need to refer to some molecular analysis to put it into a diagnostic category of RCC.
 7a Once we’ve done that, the next phrase is to determine the subtype. There are many different subtypes of RCCs really based primarily on the appearance of the tumor cells and their architectural growth patterns. Sometimes they can rely on immunohistochemical, some of the molecular properties or genetic profiles that put it in the proper subtype category.
7a Once we’ve done that, the next phrase is to determine the subtype. There are many different subtypes of RCCs really based primarily on the appearance of the tumor cells and their architectural growth patterns. Sometimes they can rely on immunohistochemical, some of the molecular properties or genetic profiles that put it in the proper subtype category.
Now the subclassification of RCCs and probably this is familiar. You’re familiar with RCCs and it is not so simple. It’s an evolving, sort of complex and ever-changing categorization. In fact, the overall categorization of subtypes just changed a few months ago. We like to think about RCC and subtypes in a sort of developmental pathway.
There is a sporadic type– that which just happened to occur–which is probably the type of cancer that most people in this room happen to have. Those are our typical clear cell, chromophobe, papillary renal cell carcinomas or maybe a few of the other rare variants.
There are those which tend to be familiar; these represent 90+ percent of all RCCs. The familial patterns–again what is associated—they are pretty rare. They are associated with and in families, multiple tumors. Different family members can have these, and we will talk a little bit more about these. There is actually going to be a talk about later in the afternoon or the morning about genetic-based or familial-based RCCs.
There are those rare—really associated with treatment of other types of cancers, and there is unusual category when you have scarred or damaged kidneys. Those kidneys are at risk for developing RCC.
Let’s move through this little bit. Once we have made the diagnosis of RCC, we’ve sub categorized it. I know it seems complex, but there are really only three or four main subtypes that we really need to talk about, especially in the context of a setting like this.
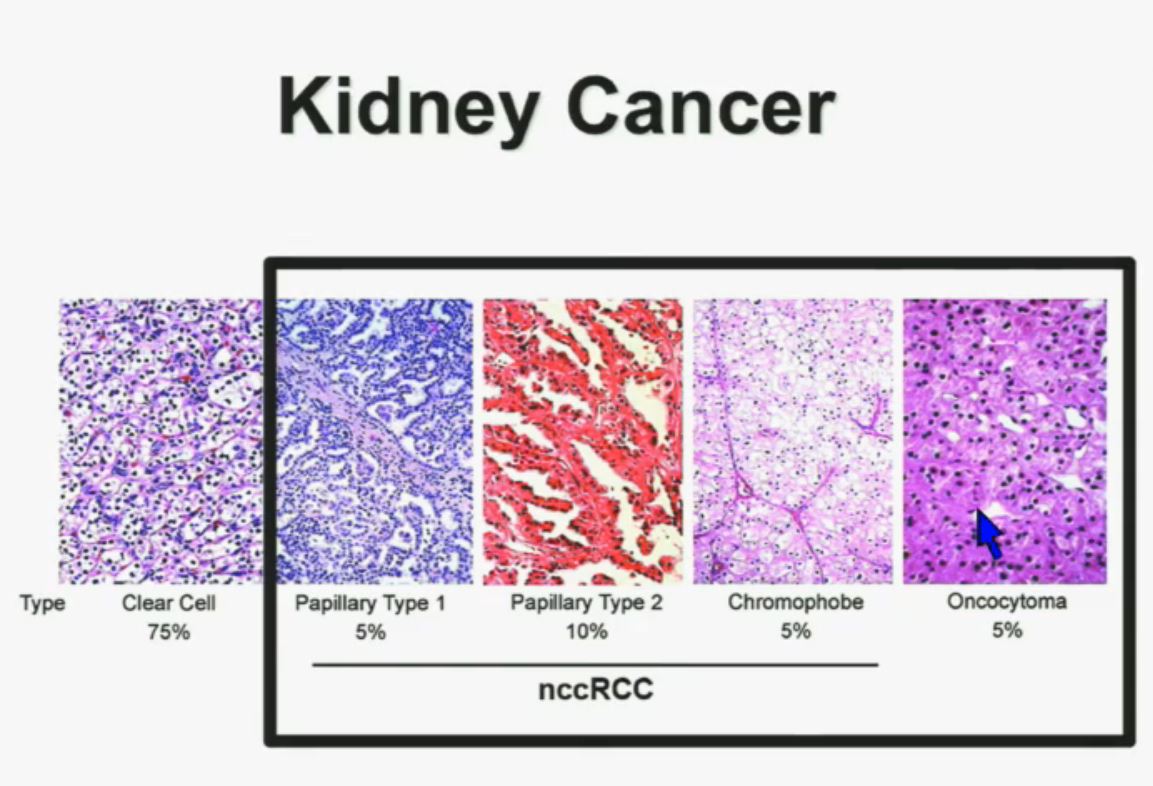
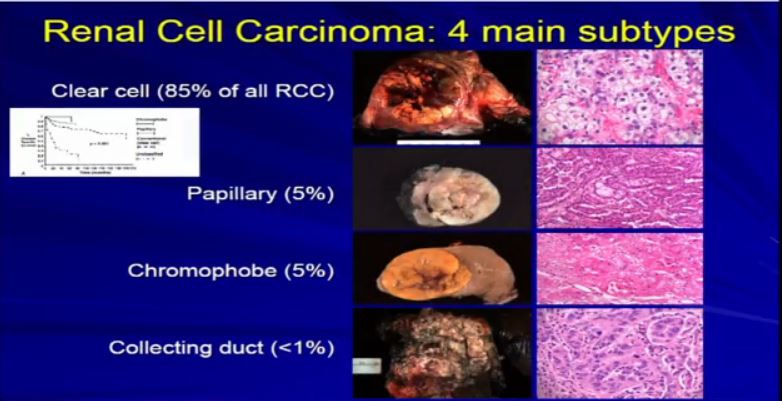
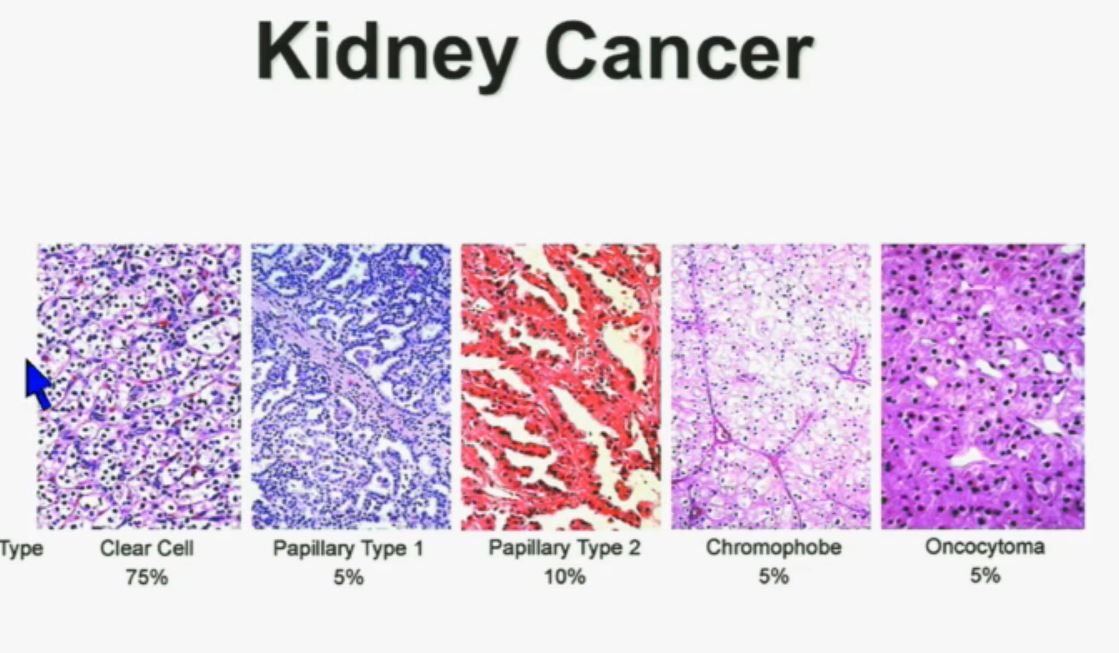
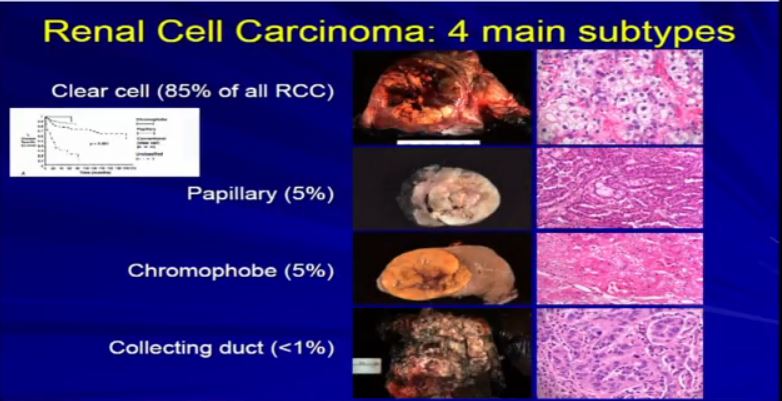 8 The most common subtype is the clear cell type. This represents about the vast majority of all sporadic types of renal cell carcinoma. Then there are the papillary and chromophobe renal cell carcinomas. Since these are really the usual types. The much less common type is collecting duct carcinoma which is really more like a urothelial cancer, it behaves like a urothelial cancer, it’s a more aggressive type of RCC.
8 The most common subtype is the clear cell type. This represents about the vast majority of all sporadic types of renal cell carcinoma. Then there are the papillary and chromophobe renal cell carcinomas. Since these are really the usual types. The much less common type is collecting duct carcinoma which is really more like a urothelial cancer, it behaves like a urothelial cancer, it’s a more aggressive type of RCC.
These are really the main four that we need to be concerned about. They are each unique based on their gross appearance and these are all partial nephrectomies (this is complete down here). Look at their gross appearance. They are very unique under the microscope. Look at their microscopic appearance.
The clear cell is clear, the papillary, very architectural pattern of a papillary tumor. These are chromophobe. This unusual eosinophilic cytoplasm are the tumor cells. Probably doesn’t mean a lot to you, but it means a lot to us, also to some other clinicians. So they have very characteristic gross, microscopic and they are very unique biochemical—and as Dr. Kim alluded to—very specific molecular and genetic profiles as well. This is all really evolving as we speak.
And we all know—this is small graph—that these also behave differently, Some behave better than others, so it is really important that we subclassify these RCCs based on their appearance—all the appearances that we talked about.
 9 The other thing that Dr. Kim alluded to, and I think we are going to talk about this a little later, and I won’t get into detail on this, but just to point out that the sub-classifications, the sub-categories, they respond differently to the different armamentaria that we have in terms of treatment modalities. So it’s very important for the pathologist to sub classify the type of RCC.
9 The other thing that Dr. Kim alluded to, and I think we are going to talk about this a little later, and I won’t get into detail on this, but just to point out that the sub-classifications, the sub-categories, they respond differently to the different armamentaria that we have in terms of treatment modalities. So it’s very important for the pathologist to sub classify the type of RCC.
 10 So on any standard pathology report, you are going to see the diagnosis, RCC, then the subtype, buried somewhere in the report; It will say, clear cell type, papillary type, chromophobe. That’s a very important part of the report.
10 So on any standard pathology report, you are going to see the diagnosis, RCC, then the subtype, buried somewhere in the report; It will say, clear cell type, papillary type, chromophobe. That’s a very important part of the report.
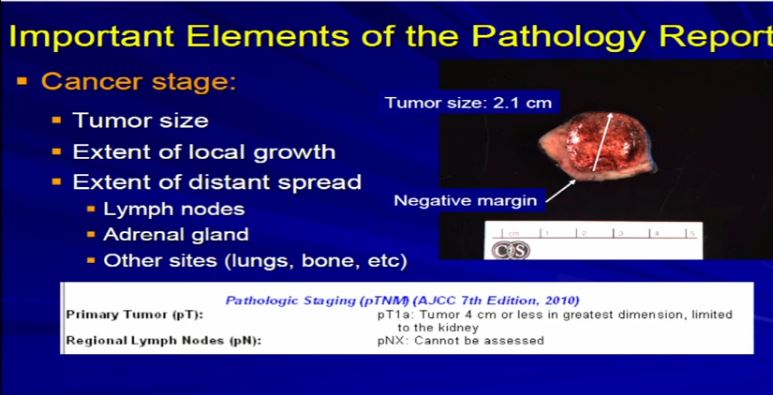 11 After diagnosis, the next important aspect is the cancer stage; The cancer stage is really defined by the size of the tumor and its local growth. Is it extending, is it staying confined to the kidney, outside the kidney to the local fat, is it going into any regional lymph nodes that might have been removed during surgery, or was it extending into the adrenal gland, which might have been removed as well? So we analyze each case on what we have and what we see.
11 After diagnosis, the next important aspect is the cancer stage; The cancer stage is really defined by the size of the tumor and its local growth. Is it extending, is it staying confined to the kidney, outside the kidney to the local fat, is it going into any regional lymph nodes that might have been removed during surgery, or was it extending into the adrenal gland, which might have been removed as well? So we analyze each case on what we have and what we see.
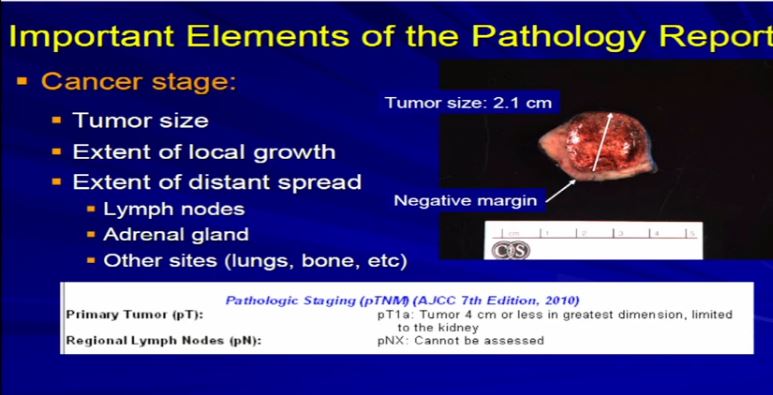
This is a typical example of a partial nephrectomy specimen of clear cell carcinoma with a margin that’s out here. Here it measures about 2.1 centimeters the margin is negative. This is a very small tumor of clear cell RCC. This would stage out at T1a, pretty low stage tumor. This would have a pretty good prognosis based on that staging profile.
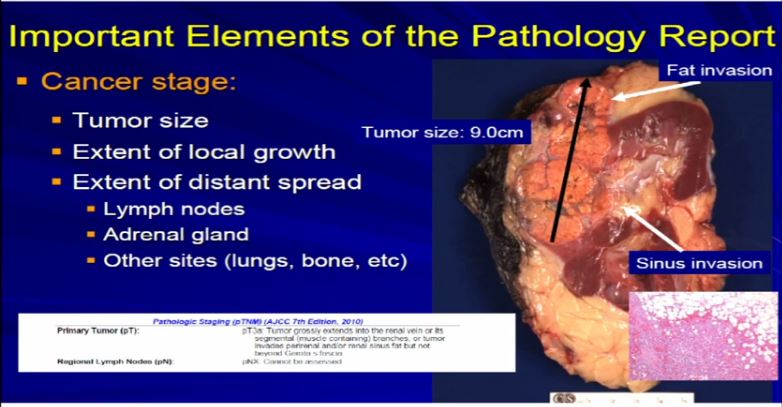
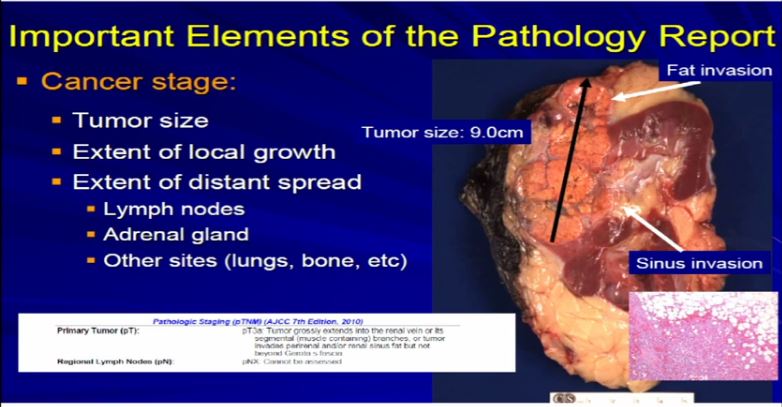 12 Now compare that with this tumor which is a complete nephrectomy specimen, shown the kidney, a lot of nephritic fat. Here’s the sinus of the kidney and here’s the tumor out here. Much bigger, about 9 centimeters and it is growing into the fat. It’s growing into the sinus fat; it is demonstrating more aggressive local growth. This would stage out—this is a microscopic showing it extending into fat. We would stage this out at T3a tumor, as it is obviously larger and more infiltrative.
12 Now compare that with this tumor which is a complete nephrectomy specimen, shown the kidney, a lot of nephritic fat. Here’s the sinus of the kidney and here’s the tumor out here. Much bigger, about 9 centimeters and it is growing into the fat. It’s growing into the sinus fat; it is demonstrating more aggressive local growth. This would stage out—this is a microscopic showing it extending into fat. We would stage this out at T3a tumor, as it is obviously larger and more infiltrative.
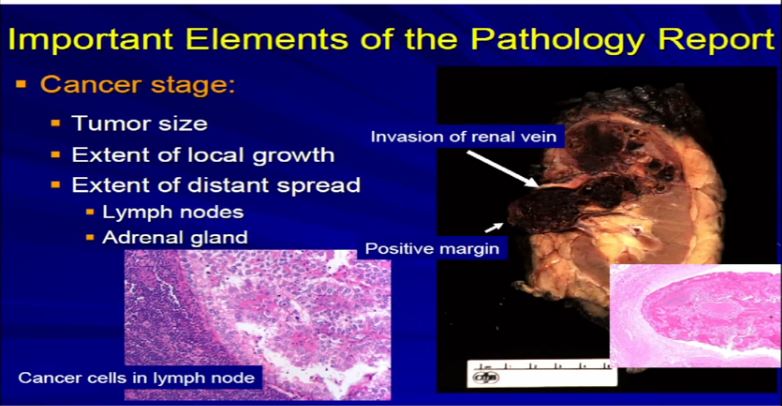
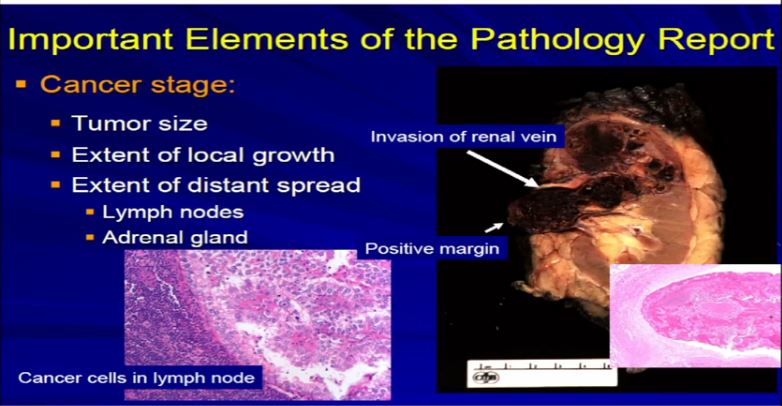 13 A different example would be the same thing. A RCC clear cell type; this is a full nephrectomy specimen. Here’s the kidney. Notice that the tumor is extending into the renal vein. This is another feature that we analyze and look for. We look for it grossly and microscopically and look for tumor extension into that vein, because that will upstage the tumor, overall tumor stage, and this is associated with generally adverse outcome. It is telling us this tumor is behaving more aggressively with local growth. We might see a lymph node, with metastatic clear cell RCC. Again, another aspect we would examine grossly and microscopically.
13 A different example would be the same thing. A RCC clear cell type; this is a full nephrectomy specimen. Here’s the kidney. Notice that the tumor is extending into the renal vein. This is another feature that we analyze and look for. We look for it grossly and microscopically and look for tumor extension into that vein, because that will upstage the tumor, overall tumor stage, and this is associated with generally adverse outcome. It is telling us this tumor is behaving more aggressively with local growth. We might see a lymph node, with metastatic clear cell RCC. Again, another aspect we would examine grossly and microscopically.
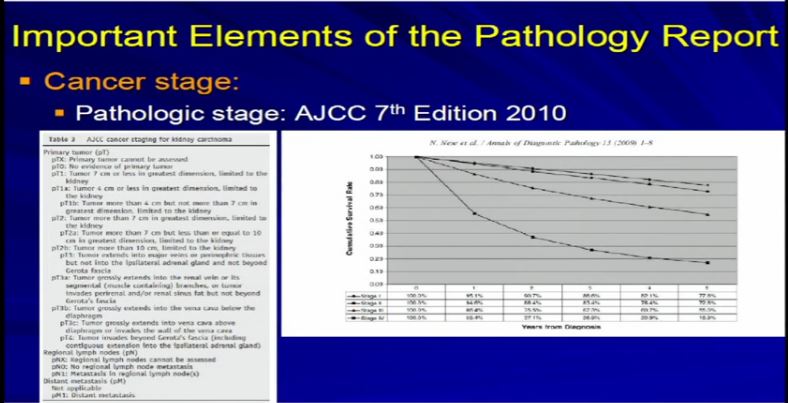
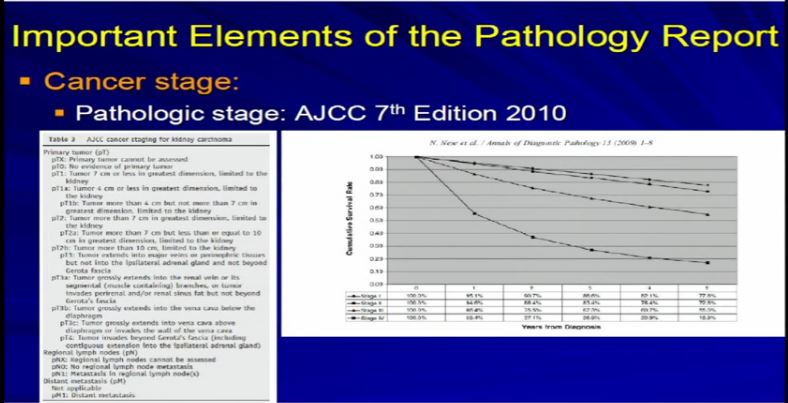 15 So we take all these features, once we have analyzed the tumor and we apply the grading system created by the Joint Council on Cancer Staging, the AJCC and we apply the pathologic stage. Why? Because as Dr. Kim alluded to, we all know, that cancer staging, and it is true for any type of cancer, the higher the stage, the more aggressive that tumor will likely behave therefore the therapy needs to be tailored to their particular stage. And the report should indicate clearly dictate the tumor stage. And that’s part of the standard reporting. Any good cancer report.
15 So we take all these features, once we have analyzed the tumor and we apply the grading system created by the Joint Council on Cancer Staging, the AJCC and we apply the pathologic stage. Why? Because as Dr. Kim alluded to, we all know, that cancer staging, and it is true for any type of cancer, the higher the stage, the more aggressive that tumor will likely behave therefore the therapy needs to be tailored to their particular stage. And the report should indicate clearly dictate the tumor stage. And that’s part of the standard reporting. Any good cancer report.
 14 The final cancer features I’m going to talk about we’re talking about are; resection margin, the grade, vascular invasion, tumor necrosis and this this unusual rhabdoid or sarcomatoid differentiation. These are elements which go beyond cancer staging and the diagnosis. Here’s two examples.
14 The final cancer features I’m going to talk about we’re talking about are; resection margin, the grade, vascular invasion, tumor necrosis and this this unusual rhabdoid or sarcomatoid differentiation. These are elements which go beyond cancer staging and the diagnosis. Here’s two examples.
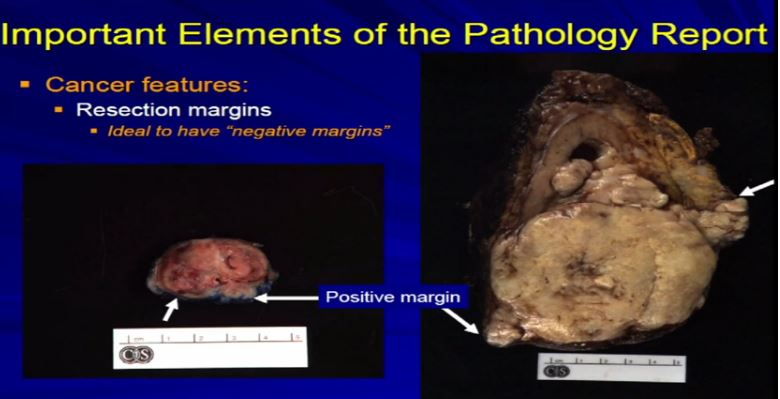
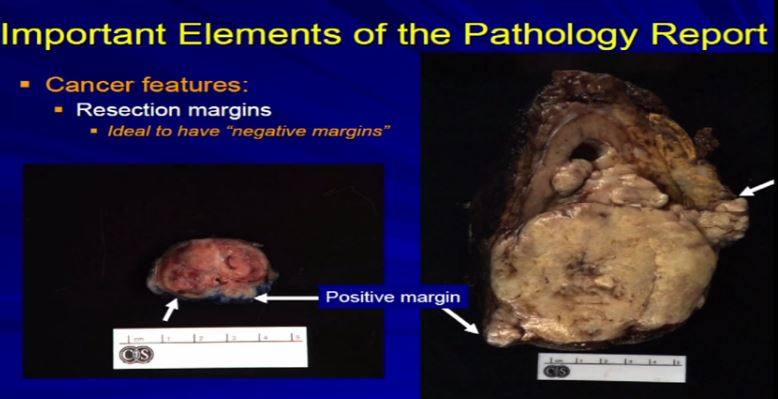 16 Let us talk about resection margins. These are indirectly related to or they indicate the local aggressiveness of a tumor, if they are growing to a margin. It’s ideal when a partial nephrectomy or a complete nephrectomy is performed, as we have here, the surgeons always try to get the whole thing out so we achieve negative margins . That is optimal. Sometimes it’s not possible, especially if we have a high stage RCC like this one which is extending into fat. Sometimes it’s impossible to get a clear margin. This might get portend some additional therapy when it comes to therapeutic– time for a therapy . With a smaller resection sometimes it’s impossible to get a negative margin or the surgeon needs to go back and take cleaner margins. That interpreted for frozen section analysis, and clear out that margin, again because optimally, we want to achieve a negative resection margin.
16 Let us talk about resection margins. These are indirectly related to or they indicate the local aggressiveness of a tumor, if they are growing to a margin. It’s ideal when a partial nephrectomy or a complete nephrectomy is performed, as we have here, the surgeons always try to get the whole thing out so we achieve negative margins . That is optimal. Sometimes it’s not possible, especially if we have a high stage RCC like this one which is extending into fat. Sometimes it’s impossible to get a clear margin. This might get portend some additional therapy when it comes to therapeutic– time for a therapy . With a smaller resection sometimes it’s impossible to get a negative margin or the surgeon needs to go back and take cleaner margins. That interpreted for frozen section analysis, and clear out that margin, again because optimally, we want to achieve a negative resection margin.
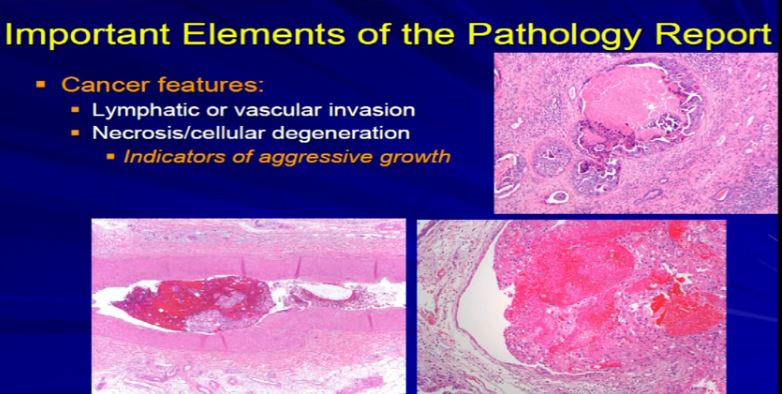
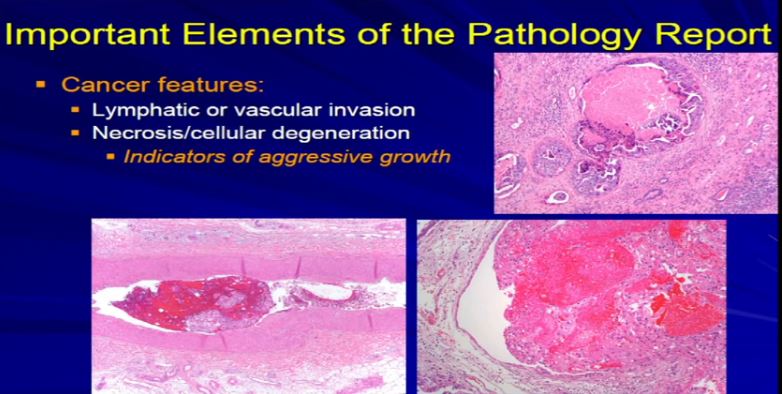 17 The next factor is vascular invasion. When the tumor invades into those lymphatics that Dr. Kim talked about in surgery. They have a propensity for them to go to the lymph node or they can go into veins or even sometimes arteries and then they have unfortunately, the capacity to go to the lungs or bones or other sites. Those confer an adverse prognostic indicator. Those are an indicator that this tumor might behave in a more aggressive manner. So if we see it microscopically, we include it in the report. Also if there’s tumor cell degeneration and necrosis, that is usually associated aggressive growth in the tumor and we will report that, too. Sometimes that will dictate how the next round of therapy will be undertaken.
17 The next factor is vascular invasion. When the tumor invades into those lymphatics that Dr. Kim talked about in surgery. They have a propensity for them to go to the lymph node or they can go into veins or even sometimes arteries and then they have unfortunately, the capacity to go to the lungs or bones or other sites. Those confer an adverse prognostic indicator. Those are an indicator that this tumor might behave in a more aggressive manner. So if we see it microscopically, we include it in the report. Also if there’s tumor cell degeneration and necrosis, that is usually associated aggressive growth in the tumor and we will report that, too. Sometimes that will dictate how the next round of therapy will be undertaken.
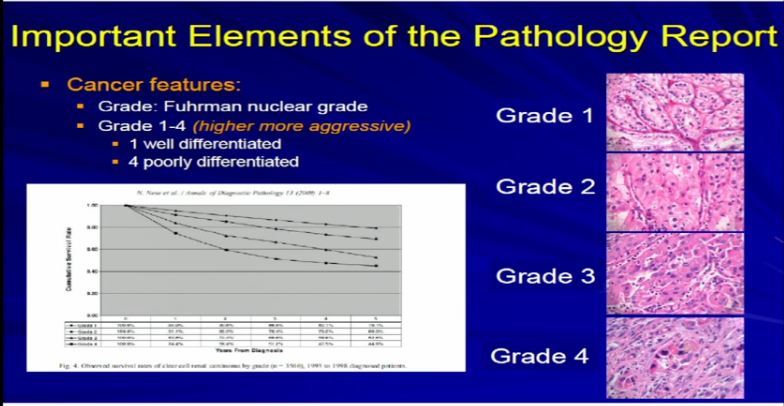 18 Dr. Kim already talked about tumor grade. We apply–the pathologist applies the tumor grade. The Fuhrman grade is the one that is used for RCC, and it a grading system for 1 to 4. Really, it delineates the degree of differentiation. Grade 1s are well-differentiated tumor, grade 4 are poorly differentiated and in any type of tumor–doesn’t matter if it’s breast, color, renal cell carcinoma–generally well-differentiated tumors behave better than poorly-differentiated tumors.And we assign a grade based on our observations.
18 Dr. Kim already talked about tumor grade. We apply–the pathologist applies the tumor grade. The Fuhrman grade is the one that is used for RCC, and it a grading system for 1 to 4. Really, it delineates the degree of differentiation. Grade 1s are well-differentiated tumor, grade 4 are poorly differentiated and in any type of tumor–doesn’t matter if it’s breast, color, renal cell carcinoma–generally well-differentiated tumors behave better than poorly-differentiated tumors.And we assign a grade based on our observations.
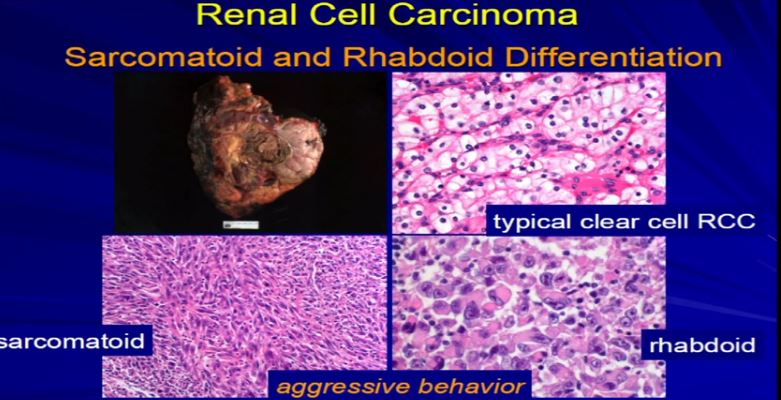 19 Finally, sarcomatoid or rhabdoid differentiation. Most tumors will have just one type of differentiation. This is an example of RCC. The vast majority are RCCclear cell, the conventional type. But in it, there were some pockets where the tumor cells had this unusual morphology under the microscope, called sarcomatoid differentiation, or over here, with we had this rhabdoid differentiation. You can see it that it’s very different than clear cell. These, for whatever reason, are associated with tumor aggressiveness. So when we see this, we need to report it. We need to quantitate it, and we put it in the report because these mandate some additional therapy, independent of stage, because they are really associated with aggressive tumors
19 Finally, sarcomatoid or rhabdoid differentiation. Most tumors will have just one type of differentiation. This is an example of RCC. The vast majority are RCCclear cell, the conventional type. But in it, there were some pockets where the tumor cells had this unusual morphology under the microscope, called sarcomatoid differentiation, or over here, with we had this rhabdoid differentiation. You can see it that it’s very different than clear cell. These, for whatever reason, are associated with tumor aggressiveness. So when we see this, we need to report it. We need to quantitate it, and we put it in the report because these mandate some additional therapy, independent of stage, because they are really associated with aggressive tumors
All these last category features that I talked about, once we have observed them, we include them in the report. Again, usually any standard RCC report will have these features included in them because they will really impact upon therapy. *See slide10
 20 Two quick categories and I will be done here.I was say a couple of words about hereditary genetic syndromes associated with RCC. This is taken out there that long list that I presented a few slides back. We all know that there are well-known, well-defined syndromes–genetic syndromes or familial syndromes that put you at increased risk from dying from other neoplasms, including RCC, notably Von Hippel Lindau, tuberous sclerosis, Birt-Hogg Dube, these sorts of things. The bottom line: as a pathologist, I can’t look at most of these tumors and say, “this is a clear cell carcinoma. It’s clearly Von Hippel-Lindau, tubersclerosis, or whatever.” All I can say is that it is clear cell carcinoma.
20 Two quick categories and I will be done here.I was say a couple of words about hereditary genetic syndromes associated with RCC. This is taken out there that long list that I presented a few slides back. We all know that there are well-known, well-defined syndromes–genetic syndromes or familial syndromes that put you at increased risk from dying from other neoplasms, including RCC, notably Von Hippel Lindau, tuberous sclerosis, Birt-Hogg Dube, these sorts of things. The bottom line: as a pathologist, I can’t look at most of these tumors and say, “this is a clear cell carcinoma. It’s clearly Von Hippel-Lindau, tubersclerosis, or whatever.” All I can say is that it is clear cell carcinoma.
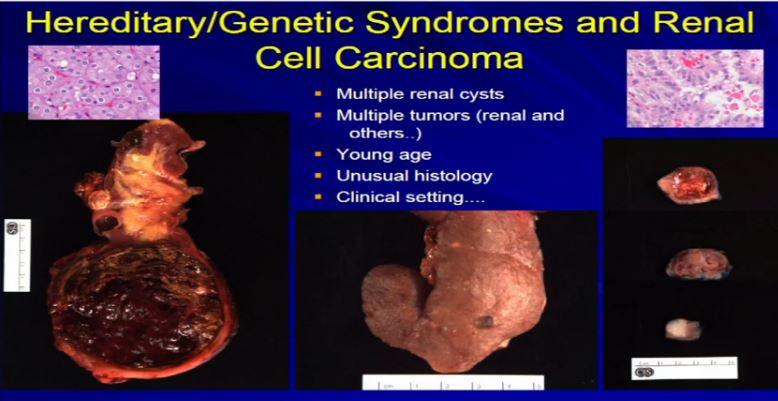
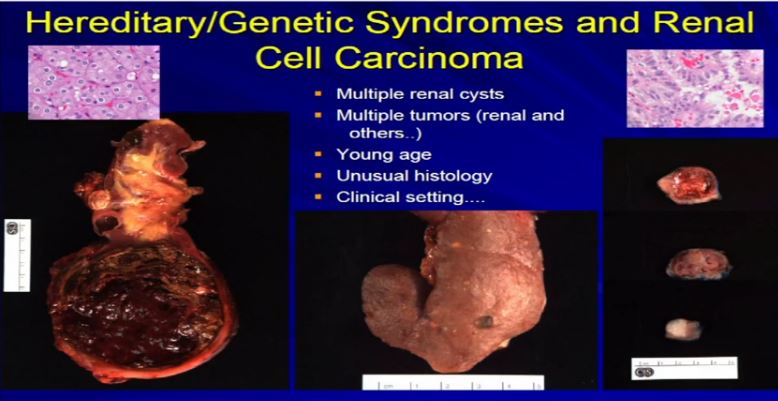 21 There are a few types of tumors that I can look at and say, if they have unusual morphology, like this tumor up here, or this tumor up here (references images) , they don’t comfortably fit into the typical types of RCC. Maybe it is a syndromic-type of carcinoma. Very, very rare, less than one percent that we would ever suggest to a clinician that maybe this is syndromic. What we can do is when we get samples like a renal resection like these three different cases, where there are multiple tumors. Here we have multiple tumors or multiple cysts—here we have maybe 20 or 30 different tumors in the particular kidney—or here’s a younger patient with one, two, three separate tumors. Then we can suggest that there is something odd about this, as we usually don’t see this in sporadic type tumors. Maybe it is associated with a genetic syndrome. So; multiple tumors, cysts, a young age, presentation of a renal cell carcinoma of unusual histology, we will suggest to your treatment team that maybe this is a genetic or syndromic pattern of RCC. There’s going to be more on this topic later this morning.
21 There are a few types of tumors that I can look at and say, if they have unusual morphology, like this tumor up here, or this tumor up here (references images) , they don’t comfortably fit into the typical types of RCC. Maybe it is a syndromic-type of carcinoma. Very, very rare, less than one percent that we would ever suggest to a clinician that maybe this is syndromic. What we can do is when we get samples like a renal resection like these three different cases, where there are multiple tumors. Here we have multiple tumors or multiple cysts—here we have maybe 20 or 30 different tumors in the particular kidney—or here’s a younger patient with one, two, three separate tumors. Then we can suggest that there is something odd about this, as we usually don’t see this in sporadic type tumors. Maybe it is associated with a genetic syndrome. So; multiple tumors, cysts, a young age, presentation of a renal cell carcinoma of unusual histology, we will suggest to your treatment team that maybe this is a genetic or syndromic pattern of RCC. There’s going to be more on this topic later this morning.
 22 The final topic I was asked to talk about the performance of secondary slide reviews. It’s kind of important. It’s really important when you come to an institution for definitive therapy, it’s always good to have that team—and we do this all the time—review the outside slides to be sure that you have an expert team who works with your treating physicians. We work as a team through tumor board reviews and discussions, and almost every discussions–. Almost every single individual case, to ensure that we have the correct diagnosis. We have the critical elements included in that report. The specific special testings have been performed, and we have accurate diagnosis and staging and things like that. What you need to do is provide, when you come here, is a copy of the reports, a set of the glass slides, sometimes we call them the recuts. That is all we need to provide an incoming secondary review.
22 The final topic I was asked to talk about the performance of secondary slide reviews. It’s kind of important. It’s really important when you come to an institution for definitive therapy, it’s always good to have that team—and we do this all the time—review the outside slides to be sure that you have an expert team who works with your treating physicians. We work as a team through tumor board reviews and discussions, and almost every discussions–. Almost every single individual case, to ensure that we have the correct diagnosis. We have the critical elements included in that report. The specific special testings have been performed, and we have accurate diagnosis and staging and things like that. What you need to do is provide, when you come here, is a copy of the reports, a set of the glass slides, sometimes we call them the recuts. That is all we need to provide an incoming secondary review.
The other scenario when you go off, you might need to off somewhere else for some additional testing for some additional therapy. In that situation, you might need to take, or you should take a set of slides with you to that institution because they will probably want to the same thing and review to ensure that we are all talking about the same disease process.
Remember that your slides or blocks, when you are treated here, or whatever institution, generally those tissue blocks are stored in an incredible huge file, either in the basement of the hospital right below us here or in a warehouse as we have done down in Torrance. T. They are basically saved forever. So when you need to go somewhere in five or ten or fifteen or twenty years, God forbid that there is a recurrence, and you need to get some additional testing, we can pull those blocks out from Torrance (CA) and create a second set of recuts, or a third or fourth set. We can send it off wherever it needs to go for some additional testing or evaluation.
 23 What you need to do is fill out this authorization form here at Cedars if you are being treated here at Cedars. All you need to do is check off “Get a copy of the pathology report” and please provide a set or recut. It’ll take a few days, three days. We’ll get that for you, send it where it needs to go, or we can give it to you directly and you can just carry it with you to that next institution or wherever you need to go.”
23 What you need to do is fill out this authorization form here at Cedars if you are being treated here at Cedars. All you need to do is check off “Get a copy of the pathology report” and please provide a set or recut. It’ll take a few days, three days. We’ll get that for you, send it where it needs to go, or we can give it to you directly and you can just carry it with you to that next institution or wherever you need to go.”
With that Dr. Luthringer thanks the KCA, the audience and Dr. Figlin for the chance to speak. And with that, I agree remind you to get a copy of your own pathology report, and know where your slides are stored. If there is any questions as to your own pathology, if the tumor seems to be unusual, or of an especially high grade, do yourself and your family a big favor, and discuss whether a review of your slides is in order!
With this rare disease, and the complexity of doing the kind of analysis you see here, do not be afraid to get that second opinion. Go back and see so that pathology may affect the treatment options given–very important!




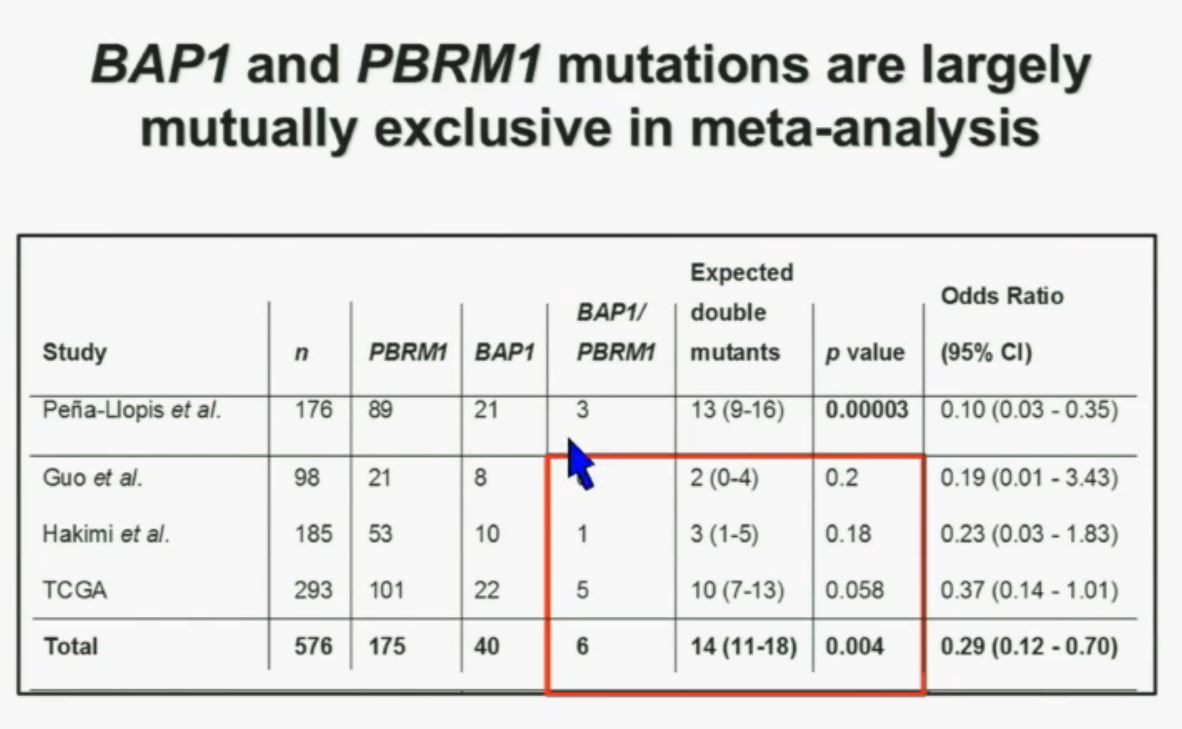

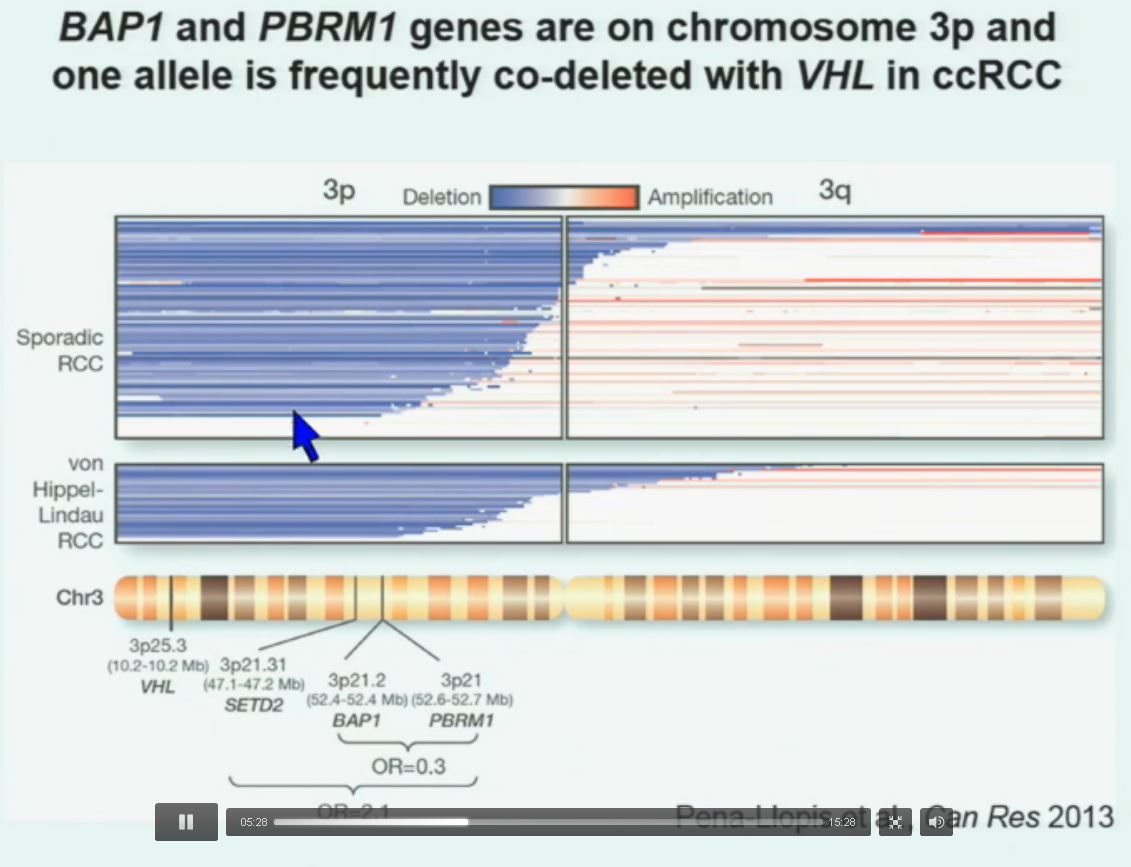
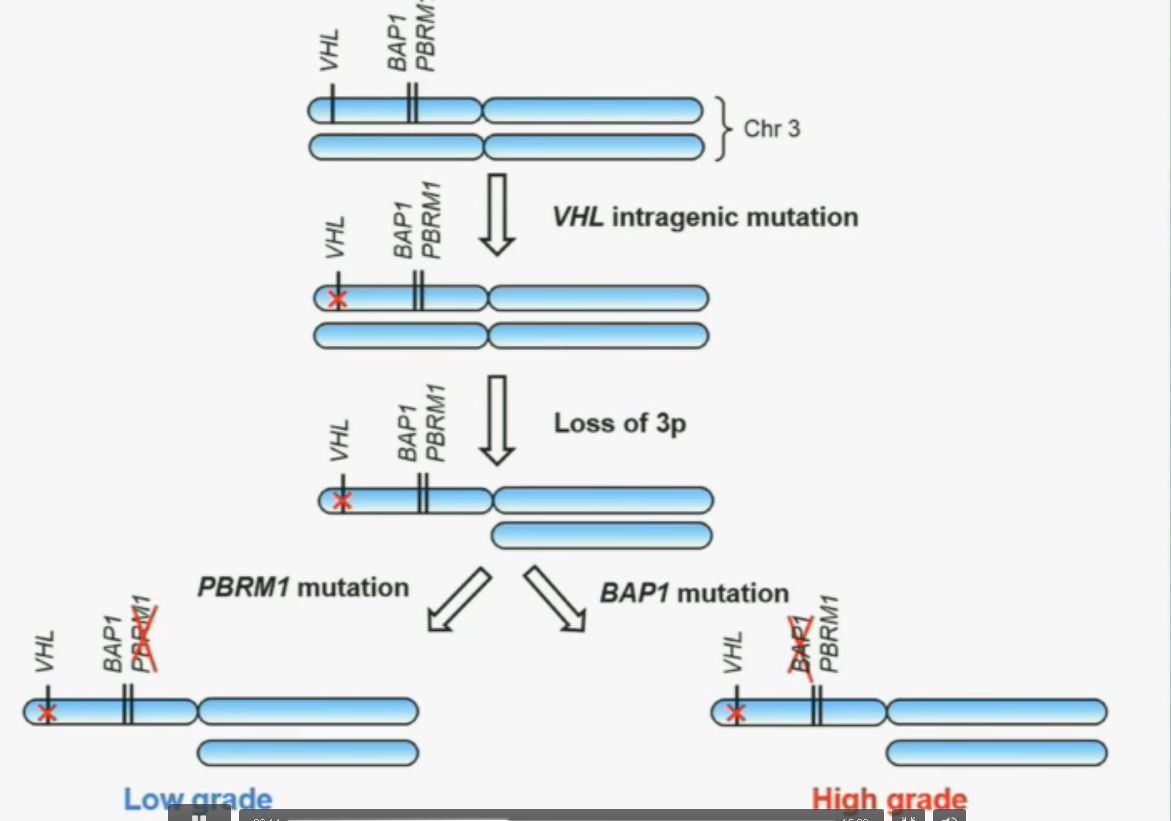
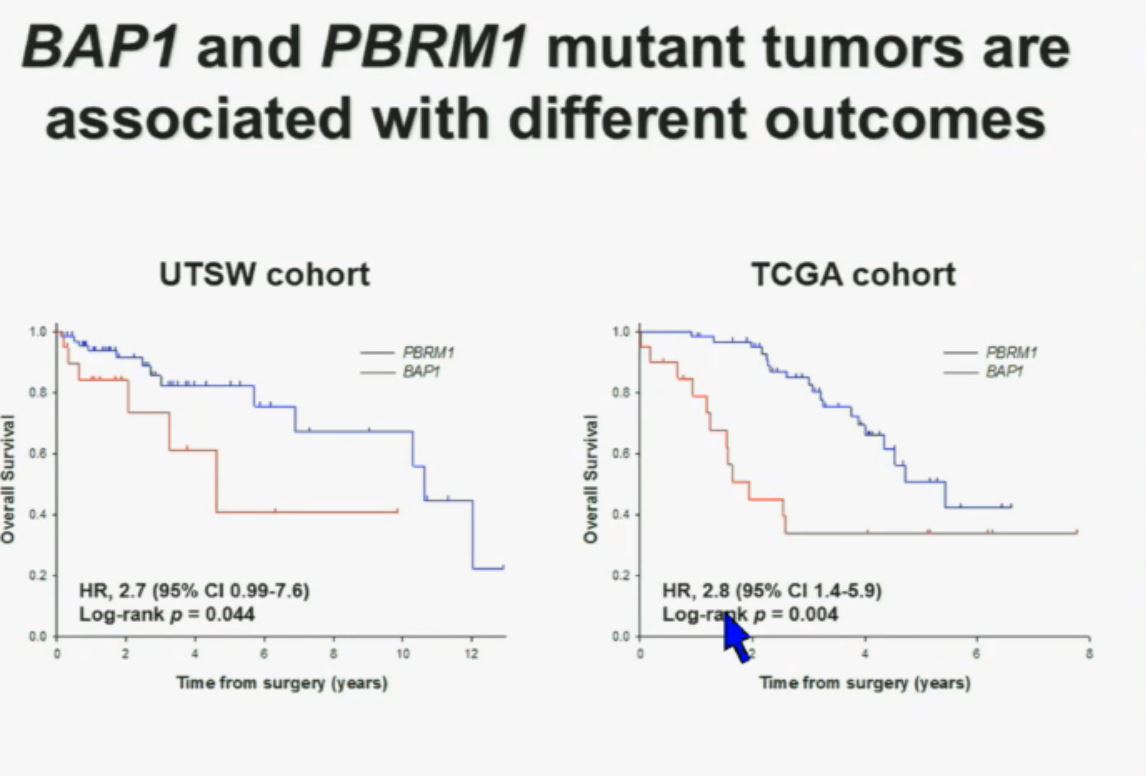

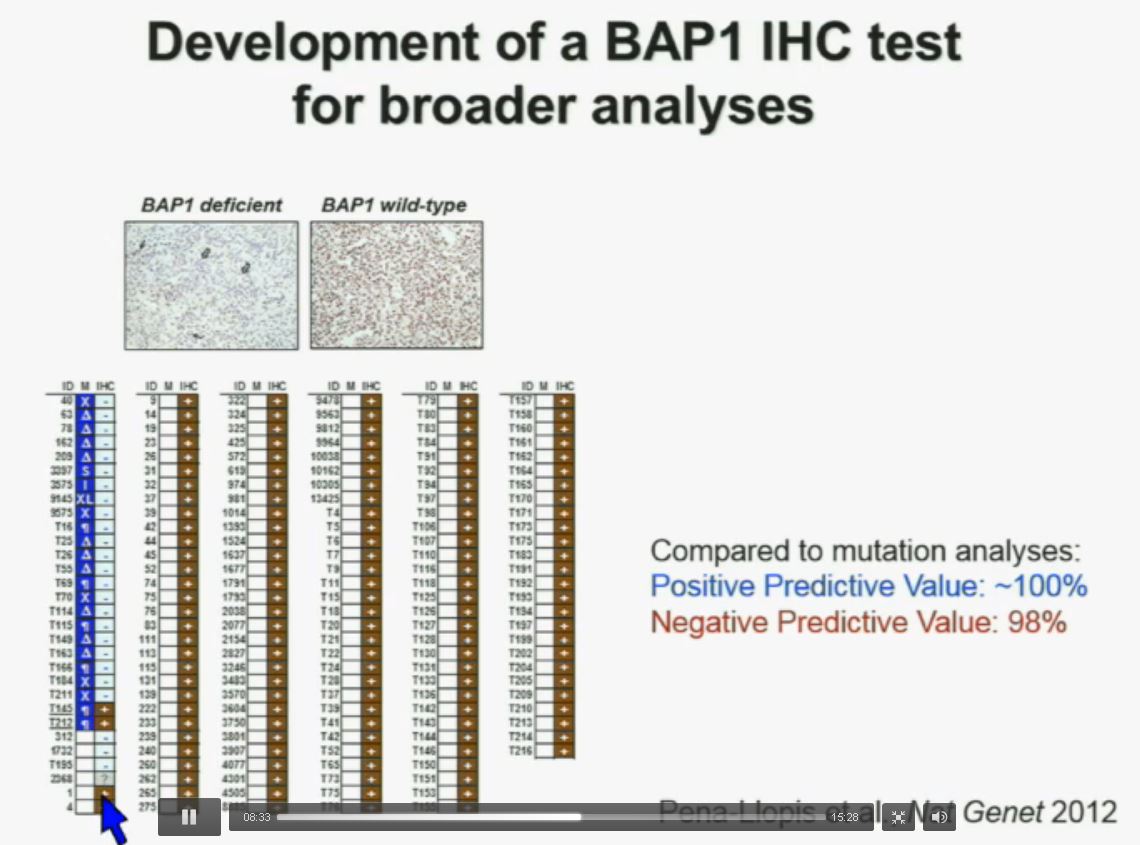
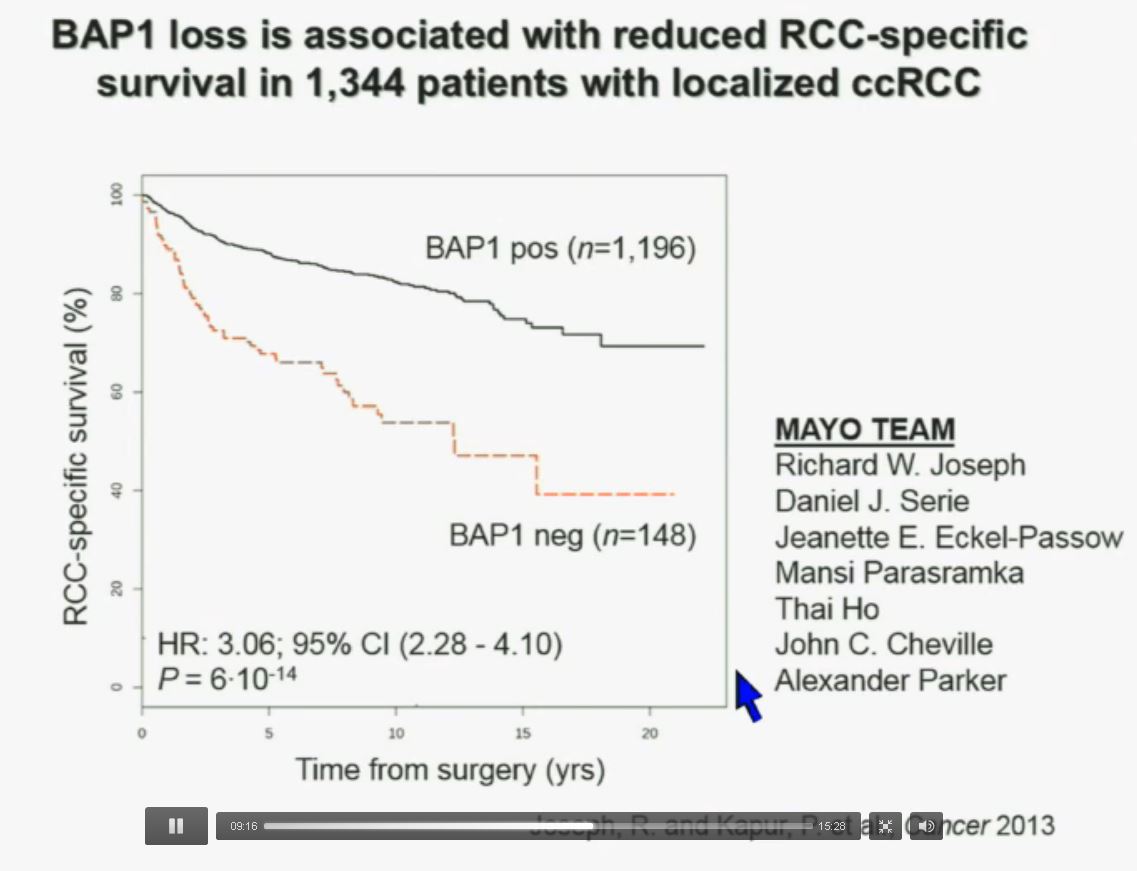
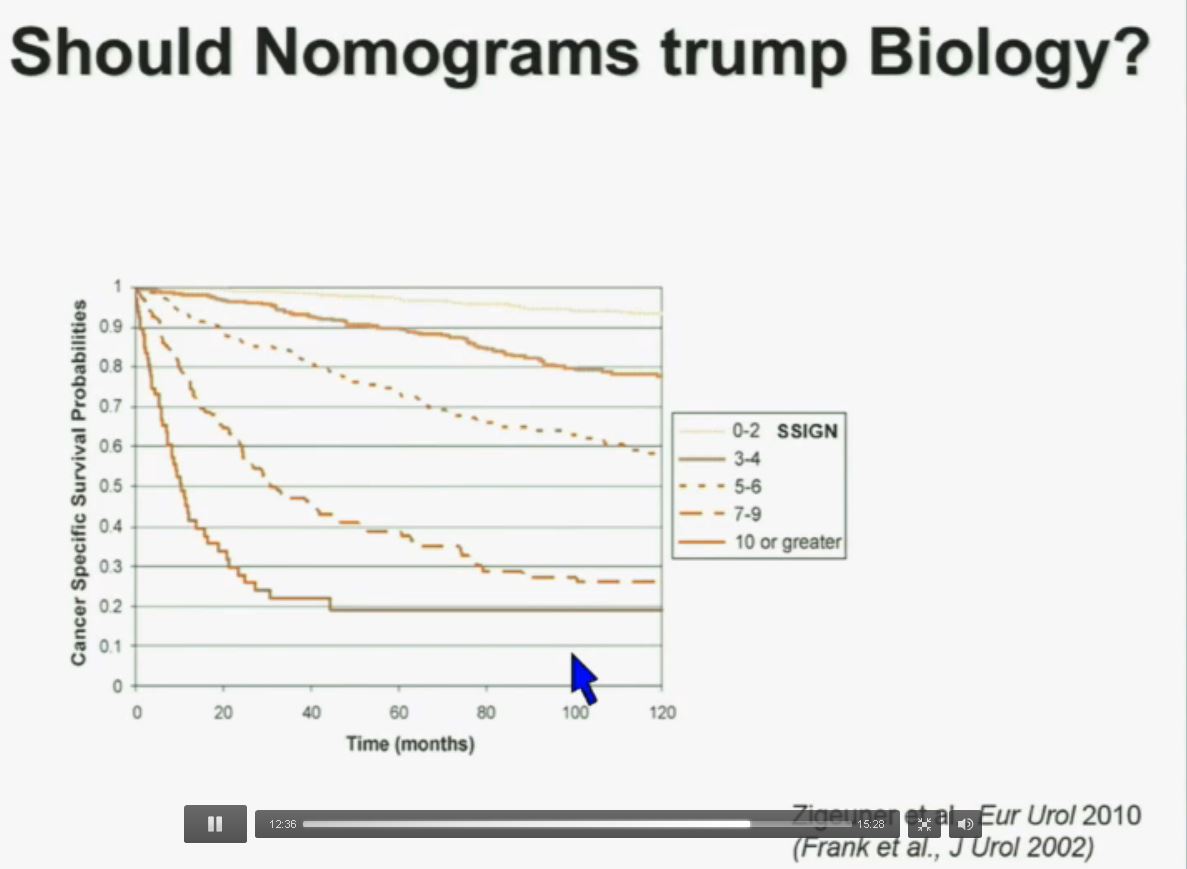
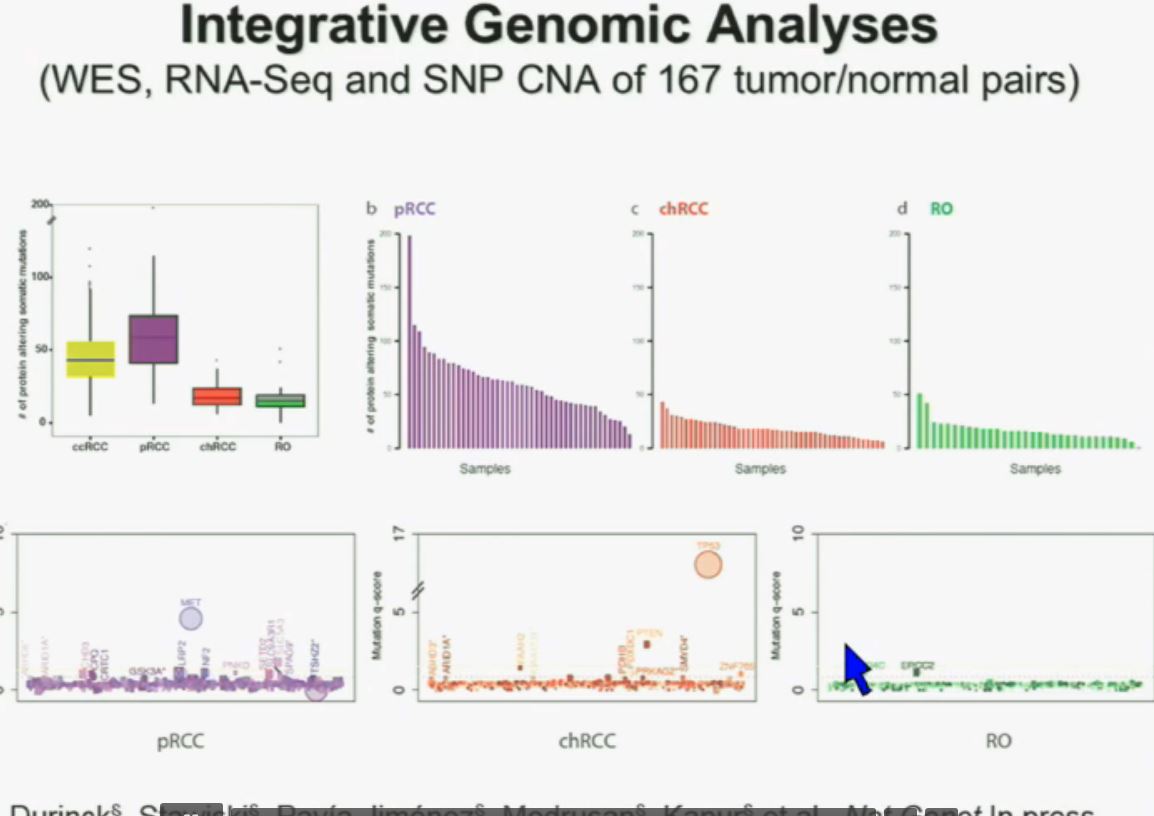
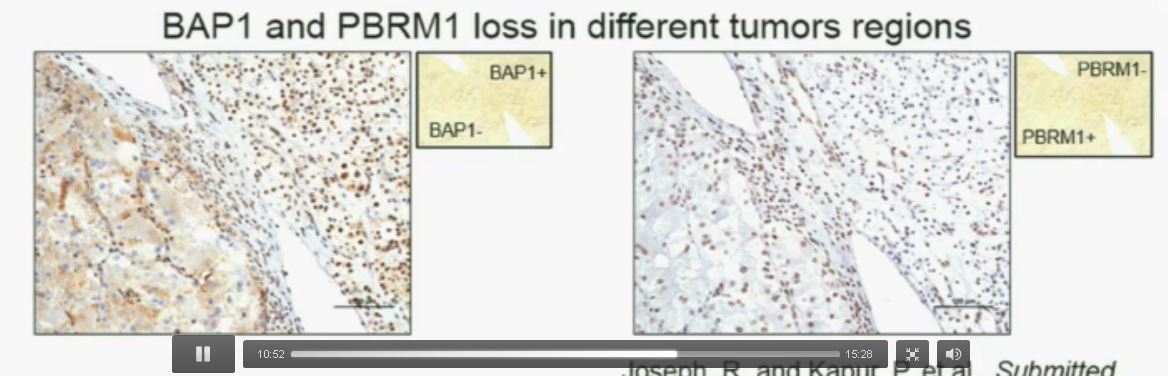
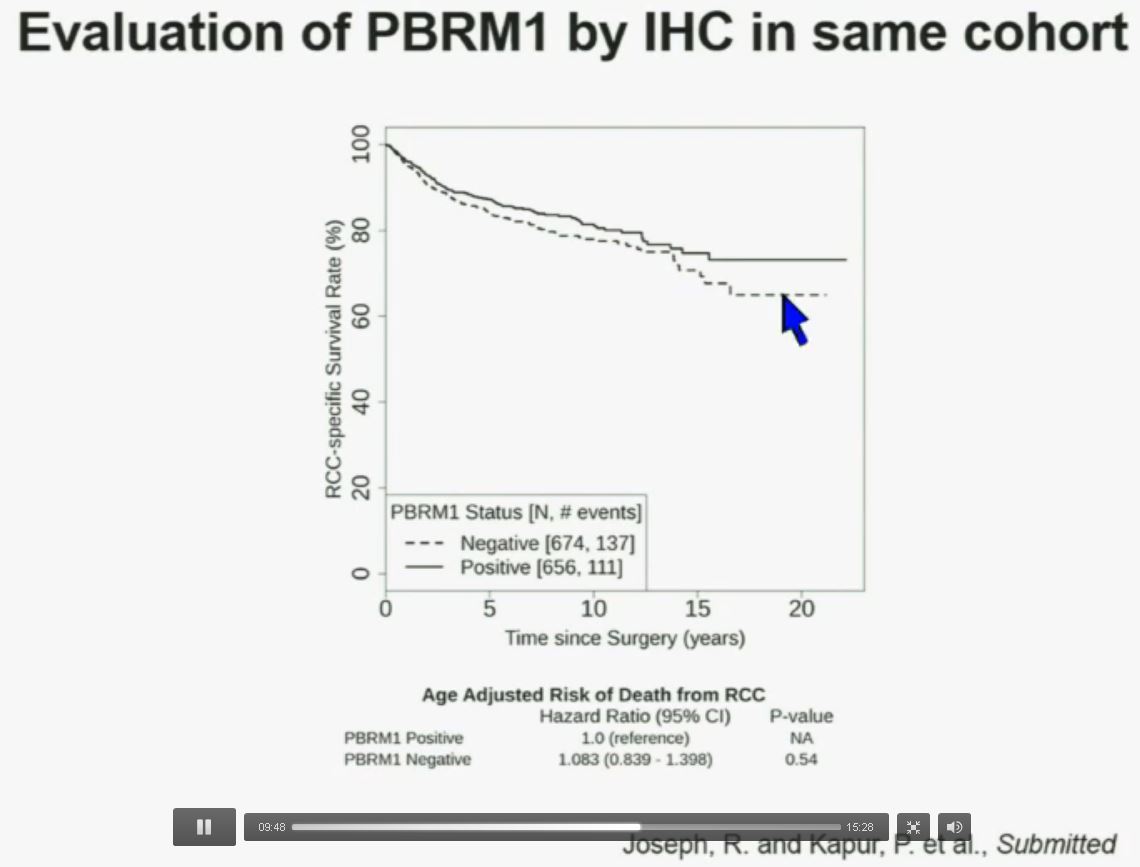 Now in the same cohort we looked at BPRM1, which like BAP1 in a two-hit tumor suppressor gene, and we find no significant differences.
Now in the same cohort we looked at BPRM1, which like BAP1 in a two-hit tumor suppressor gene, and we find no significant differences.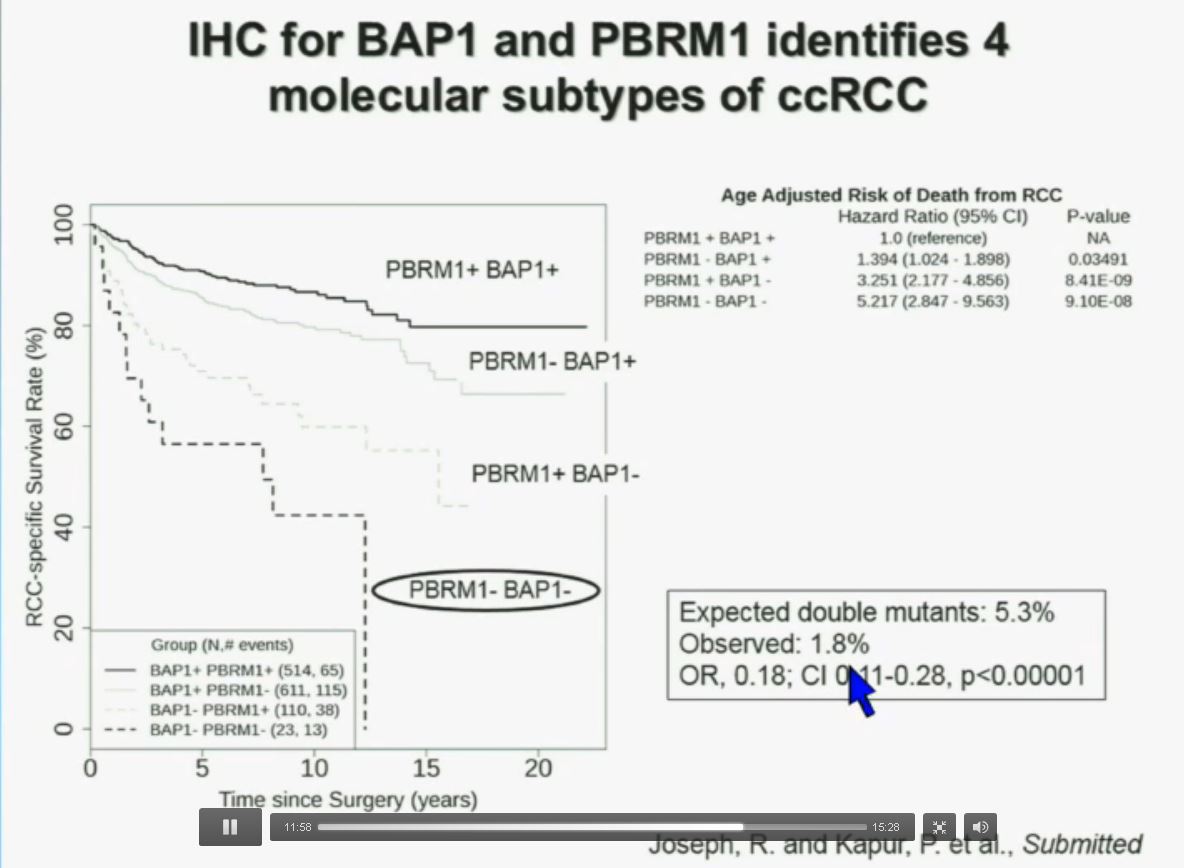
 Slide C
Slide C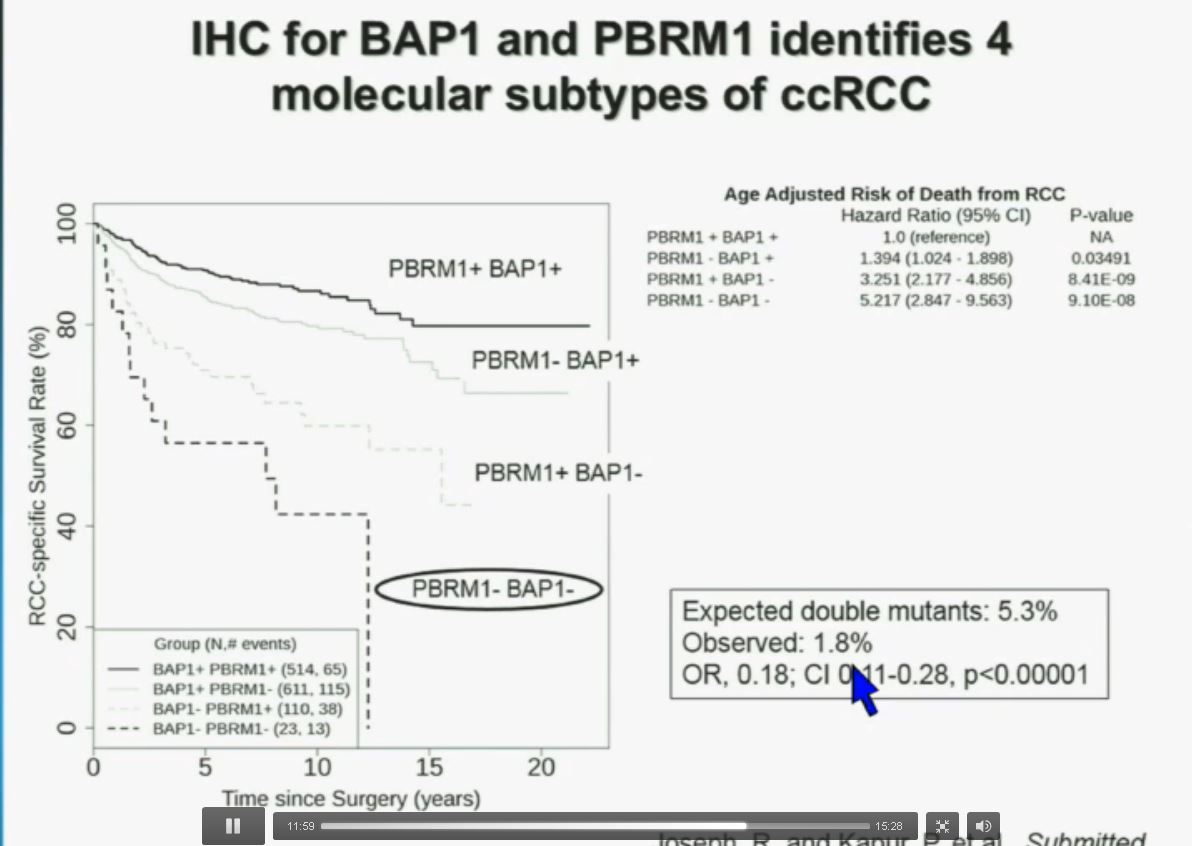 Slide should include quote “BAP1 and PBRM1 do NOT predict outcomes independently of SSIGN”
Slide should include quote “BAP1 and PBRM1 do NOT predict outcomes independently of SSIGN”